WO2017168174A1 - New pharmaceutical forms of sildenafil - Google Patents
New pharmaceutical forms of sildenafil Download PDFInfo
- Publication number
- WO2017168174A1 WO2017168174A1 PCT/GB2017/050921 GB2017050921W WO2017168174A1 WO 2017168174 A1 WO2017168174 A1 WO 2017168174A1 GB 2017050921 W GB2017050921 W GB 2017050921W WO 2017168174 A1 WO2017168174 A1 WO 2017168174A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sildenafil
- component
- pharmaceutical composition
- composition according
- crystal
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0002—Galenical forms characterised by the drug release technique; Application systems commanded by energy
- A61K9/0004—Osmotic delivery systems; Sustained release driven by osmosis, thermal energy or gas
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0002—Galenical forms characterised by the drug release technique; Application systems commanded by energy
- A61K9/0007—Effervescent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
Definitions
- the present invention relates to novel forms and formulations of sildenafil.
- the present invention relates to the use of such forms and formulations of sildenafil in the treatment of male sexual dysfunction.
- Sildenafil is generically described in US patent 5,250,534 as a selective cGMP PDE inhibitor useful in the treatment of cardiovascular disorders.
- Sildenafil free base i.e. 5- ⁇ 2-ethoxy-5-[(4-methylpiperazin-l -yl)sulfonyl]phenyl ⁇ -l- methyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, is specifically described in US patent 6,204,383 Bl as an agent with pharmaceutical utility in the treatment of male sexual dysfunction.
- Sildenafil free base has the following structure:
- phenol co-crystals improve solubility profile of sildenafil to bring about rapid onset of action.
- the phenol co-crystals improve solubility at all levels of pH therefore improving the performance of the drug during fasted and non- fasted stomach condi- tions.
- Example 1 illustrates the preparation of solid sildenafil by the evaporation of a solution in a mixture of dichloromethane and methanol.
- Examples 2 and 6 describe a solution of sildenafil iodide in dichloromethane, but this is not isolated as a solid product.
- sildenafil citrate The citrate salt of sildenafil (sildenafil citrate) is currently marketed as Viagra® in numerous countries including the USA and the European Union.
- Sildenafil is the active ingredient in Viagra® which is a potent inhibitor of PDE receptors in particular the subtype 5 which is associated with male erectile dysfunction. Sildenafil is a potent muscle relaxant associated with the arterial walls in the cardio vascular system. Sildenafil also antagonises other PDE sub types such as PDE 2 and 6 which are associated with cardiac contractility and blue haze respectively. Whilst Viagra is a very potent and selective drug its pharmacological profile is far from ideal.
- a first step in solving this problem is finding variants of the product, i.e. rapidity longevity and maintenance within the therapeutic window from dose to dose which possess physical and chemical characteristics which are conclusive to addressing one or more of the problems associated with PDE 5 inhibition such as rapidity of action duration of effect and sustenance within the therapeutic window for prolonged periods of time from dose to dose.
- Co-crystals of many substances are known, co-crystals impart properties of the co- crystal partner (co-crystal former, or co-former) on the target molecule.
- co-crystal former or co-former
- Sildenafil is commercially available as the citrate salt and is therefore a good choice as a starting material.
- Other salts of sildenafil have been disclosed in the art however none have been tested for rapidity or duration of effect or for any tendency to be within the therapeutic window from dose to dose.
- the present invention relates to various aspects.
- the invention provides co-crystals of sildenafil with a group of phenolic compounds.
- the invention provides a co-crystal, which co-crystal comprises sildenafil and a co-crystal former, which co-crystal former is a compound which comprises a phenol moiety.
- the co-crystal former may be a compound of formula (I) as defined herein.
- Another aspect relates to fatty acid salts of sildenafil optionally in the form of co- crystals.
- the invention provides a salt of sildenafil with a long chain fatty acid.
- the salt may be amorphous or crystalline.
- the long chain fatty acid typically has formula R-C(0)OH wherein R is Ce-24 alkyl or Ce-24 alkenyl.
- Another aspect relates to a pharmaceutical composition
- a pharmaceutical composition comprising sildenafil and/or a pharmaceutically acceptable salt or co-crystal thereof admixed with excipients in a multicom- ponent pharmaceutical composition, wherein a first component is adapted to deliver sildenafil rapidly to promote fast onset of action, and a further component is adapted to deliver the sildenafil from dose to dose wherein the sildenafil is delivered from dose to dose within the therapeutic window.
- the pharmaceutical composition of the invention may comprise sildenafil and/or a pharmaceutically acceptable salt or co-crystal thereof admixed with excipients in a multicom- ponent pharmaceutical composition, wherein a first component is adapted to deliver sildenafil rapidly to promote fast onset of action, a second component is adapted to deliver the sildenafil from dose to dose, and a third component wherein the sildenafil is delivered from dose to dose within the therapeutic window.
- the pharmaceutical composition of the invention comprises a first component which is adapted to deliver the sildenafil rapidly to promote a fast onset of action, and a second component which is adapted to deliver the sildenafil from dose to dose, and which is further adapted to deliver the active ingredient from dose to dose within the therapeutic window.
- the pharmaceutical composition may be a dosage form, typically a solid dosage form, for instance a swallow tablet.
- the invention further provides a pharmaceutical composition comprising sildenafil or a pharmaceutically acceptable salt thereof, which may be as further defined herein (for in- stance, as in the immediately preceding paragraph) wherein the pharmaceutical composition is in the form of a swallow tablet.
- the dosage form for instance a tablet (swallow tablet)
- a tablet squeeze tablet
- the dosage form comprises two components:
- first component which is adapted to deliver the sildenafil rapidly to promote a fast onset of action
- second component which is adapted to deliver the sildenafil from dose to dose, and which is further adapted also to deliver the active ingredient from dose to dose within the therapeutic window.
- the first component typically comprises from 10% to 30% by weight, for instance 20% by weight, of the total amount of sildenafil in the composition.
- the balance of sildenafil is present in the one or more further components.
- the balance of sildenafil may be present in a second component, or where second and third components are present, the balance of sildenafil is usually distributed between the second and third components.
- the total amount of sildenafil in the pharmaceutical compositions described herein (including in the dosage forms and swallow tablets), is often lOOmg, 50mg or 25 mg.
- the first component typically comprises from 10% to 30% by weight, for instance 20% by weight, of the total amount of sildenafil in the first and second components
- the second component typically comprises from 90% to 70% by weight, for instance 80% by weight, of the total amount of sildenafil in the first and second components.
- the percentage by weight refers to the percentage by weight of sildenafil free base which the particular form of sildenafil provides (i.e. weights are quoted on a "free base" basis).
- the total amount of sildenafil in the first and second components (which again refers to the total amount of sildenafil free base which the particular form or forms of sildenafil in the first and second components provide) is 100 mg, and the first component comprises from lOmg to 30mg of sildenafil and the second component comprises from 90mg to 70mg of sildenafil.
- the first component may for instance comprise 20mg of sildenafil and the second component may comprise 80mg of sildenafil.
- the total amount of sildenafil in the first and second components is 50 mg, and the first component comprises from 5mg to 15mg of sildenafil and the second component comprises from 45mg to 35mg of sildenafil.
- the first component may for instance comprise 1 Omg of sildenafil and the second component may comprise 40mg of sildenafil.
- the total amount of sildenafil in the first and second components is 25 mg, and the first component comprises from 2.5mg to 7.5mg of sildenafil and the second component comprises from 17.5mg to 22.5mg of sildenafil.
- the first component may for instance comprise 5mg of sildenafil and the second component may comprise 20mg of sildenafil.
- the sildenafil in the first component may be sildenafil free base.
- the sildenafil in the first component may be a pharmaceutically acceptable salt of sildenafil. It may for instance be sildenafil citrate. Alternatively, it may be a fatty acid salt of sildenafil.
- the sildenafil in the first component may be in the form of a co-crystal of sildenafil.
- the dosage of sildenafil quoted on a free base basis, may be as defined above.
- the sildenafil in the second component is often a pharmaceutically acceptable salt of sildenafil.
- Sildenafil citrate is one preferred choice for the second component.
- the sildenafil in the second component may be a fatty acid salt of sildenafil.
- the sildenafil in the second component is sildenafil free base.
- the sildenafil in the second component is in the form of a co-crystal of sildenafil, for instance a co-crystal of sildenafil with a compound which comprises a phenol moiety.
- the dosage of sildenafil quoted on a free base basis, may be as defined above.
- the first component typically comprises from lOmg to 35mg, for instance from lOmg to 30mg, or for instance from 20mg to 25mg, of sildenafil.
- the first component may for instance comprise 20mg of sildenafil.
- the first component may alternatively comprise 25 mg of sildenafil.
- the sildenafil in the first component may be sildenafil free base.
- the sildenafil in the first component may be a pharmaceutically acceptable salt of sildenafil. It may for instance be sildenafil citrate. Alternatively, it may be a fatty acid salt of sildenafil.
- the sildenafil in the first component may be in the form of a co-crystal of sildenafil.
- the dosage of sildenafil quoted on a free base basis, may be as defined above.
- the second component typically comprises from 40mg to 1 OOmg of sildenafil, for in- stance from 40mg to 90mg of sildenafil, or, for example, from 50 mg to 80 mg of sildenafil.
- the second component may for instance comprise 80mg of sildenafil.
- the second component may alternatively comprise 50mg of sildenafil.
- the second component in some embodiments comprises from 70mg to 90mg of sildenafil, for example 80mg of sildenafil.
- the sildenafil in the second component is often a pharmaceutically acceptable salt of sildenafil. Sildenafil cit- rate is one preferred choice for the second component.
- the sildenafil in the second component may be a fatty acid salt of sildenafil.
- the sildenafil in the second component is sildenafil free base.
- the sildenafil in the second component is in the form of a co-crystal of sildenafil, for instance a co-crystal of sildenafil with a compound which comprises a phenol moiety.
- the dosage of sildenafil quoted on a free base basis, may be as defined above.
- the first component may comprise from lOmg to 35mg of sildenafil and the second component may comprise from 40mg to lOOmg of sildenafil.
- the first component may for instance comprise from lOmg to 30mg of sildenafil, and the second component may comprise from 40mg to 90mg of sildenafil.
- the first component may for instance comprise from 20mg to 25mg of sildenafil, and the second component may comprise from 50mg to 80mg of sildenafil.
- the first component may comprise about 20mg of sildenafil and the second compo- nent may comprise about 80mg sildenafil.
- the first component may comprise about 25mg of sildenafil and the second component may comprise about 50mg sildenafil.
- the first component may be adapted to deliver sildenafil rapidly, wherein the adaptation is that the first component comprises sildenafil admixed with excipients in a manner which promotes rapid, preferably immediate, release of the sildenafil.
- the first component may comprise sildenafil admixed with excipients, which may be conventional excipients, which promote immediate release.
- the first component may for instance comprise from lOmg to 35mg, for example from lOmg to 30mg, or from 20mg to 25 mg, of sildenafil with such excipients which promote immediate release. It may for instance comprise 20mg, or 25mg, of sildenafil with such excipients which promote immediate release.
- the excipients may be conventional excipients.
- the sildenafil in the first component may be sildenafil free base.
- the sildenafil in the first component may be a pharmaceutically acceptable salt of sildenafil. It may for instance be sildenafil citrate. Alternatively, it may be a fatty acid salt of sildenafil.
- the sildenafil in the first component may be in the form of a co-crystal of sildenafil.
- the dosage of sildenafil quoted on a free base basis, may be as defined above.
- the second component of the pharmaceutical composition of the invention may comprise sildenafil with excipients, for instance conventional excipients, which promote modified release.
- the second component may for instance comprise from 40mg to lOOmg, for example from 40mg to 90mg, or from 50mg to 80mg, of sildenafil with such excipients which promote modified release. It may for instance comprise 50mg, or 80mg, of sildenafil with such excipients.
- the second component may for instance comprise from 70mg to 90mg, of sildenafil, for instance 80mg sildenafil citrate, with such excipients.
- the excipients may be conventional ex- cipients.
- the excipients which promote modified release in the second component may be polymers.
- the sildenafil in the second component is often a pharmaceutically acceptable salt of sildenafil.
- Sildenafil citrate is one preferred choice for the second component.
- the sildenafil in the second component may be a fatty acid salt of sildenafil.
- the sildenafil in the second component is sildenafil free base.
- the sildenafil in the second component is in the form of a co-crystal of sildenafil, for instance a co-crystal of sildenafil with a compound which comprises a phenol moiety.
- the dosage of sildenafil quoted on a free base basis, may be as defined above.
- sildenafil adapted in a multi component dosage form, such as a tablet, said dosage form being adapted in a first way to provide rapid re- lease of sildenafil into the bloodstream, said dosage form being adapted in a second way to further provide a maintenance dose of sildenafil within the therapeutic window, and the dosage form being adapted in a third way to provide a modified or delayed release format of the sildenafil product which lasts from dose to dose.
- each component is dependent on the choice of formulation but in general the choice of each component will be made so that the amount of active ingredient in each component delivers the right amount of drug product to ensure rapidity, longevity from dose to dose and maintenance of dose within the therapeutic window.
- the total amount of sildenafil in a formulation is at least 20mg and usually 50mg to lOOmg.
- a preferred aspect of the invention is a product (usually a multi component dosage form, such as a tablet) which comprises:
- sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co- crystal is optionally as further defined herein;
- a fatty acid salt of sildenafil suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and a delayed release component, comprising a bolus dose of sildenafil.
- the sildenafil in the bolus dose is in the form of sildenafil citrate.
- a product usually a multi component dosage form, such as a tablet
- a product which comprises:
- sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co-crystal is optionally as further defined herein;
- sildenafil in the form of a fatty acid salt of sildenafil as defined herein, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component;
- said delayed release component comprising a bolus dose of sildenafil.
- the bolus dose of sildenafil may be from 20mg to 95mg sildenafil.
- the sildenafil in the bolus dose may be in the form of sildenafil citrate.
- a product usually a multi component dosage form, such as a tablet
- a product which comprises:
- sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co-crystal is optionally as further defined herein; from 2.5 to 15mg sildenafil in the form of a fatty acid salt of sildenafil as defined herein, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and
- said delayed release component comprising a bolus dose of sildenafil.
- the sildenafil in the bolus dose may be in the form of sildenafil citrate.
- the bolus dose may for example be from lOmg to 80mg sildenafil, for instance 20mg sildenafil.
- Another preferred aspect of the invention is a product (usually a multi component dosage form, such as a tablet) which contains: 5mg of sildenafil co- crystals with a phenol to deliver rapidity, 15mg of sildenafil fatty acid adapted using time release technology to deliver sildenafil between onset of action and kick in of a bolus amount of delayed release component, and a bolus amount of delayed release component of sildenafil which is suitably 20mg.
- sildenafil refers to 80mg sildenafil free base.
- 80mg sildenafil refers to 80mg sildenafil free base.
- the sildenafil is in the form of the citrate salt, the term "80mg sildenafil” as used herein means 112mg of sildenafil citrate (if the mass of the sildenafil citrate is rounded to zero decimal places).
- the term "20mg sildenafil refers to 20mg sildenafil free base.
- sildenafil is in the form of the citrate salt
- the term "20mg sildenafil” as used herein means 28 mg of sildenafil citrate (if the mass of sildenafil citrate is rounded to zero decimal places).
- Fig. 1 shows powder X-ray diffractograms of the initial batch of sildenafil (P42) as received (upper diffractogram) and the published P42 form: QEGTUT anhydrous form (lower diffractogram).
- Fig. 2 shows powder X-ray diffractograms of the initial P42 batch as received (upper diffractogram) and after grinding (lower diffractogram).
- Fig. 3 shows powder X-ray diffractograms of P42 polymorphs P42 Form I (upper diffractogram) and P42 Form II (lower diffractogram).
- Fig. 4 shows powder X-ray diffractograms of P42 polymorphs P42-A (first and top diffractogram), P42-B (second diffractogram), P42-C (third diffractogram), P42-D (fourth diffractogram) and P42-E (fifth and bottom diffractogram).
- Fig. 5 is a diagram summarising the different behaviours of forms of P42.
- Fig. 6 shows powder X-ray diffractograms of the following new multicomponent forms of P42: P42-I-A (first and top diffractogram), P42-I-B (second diffractogram), P42-III (third diffractogram), P42-IV-A (fourth diffractogram), P42-IV-B (fifth diffractogram), P42- V-A (sixth and bottom diffractogram).
- P42-VI-B first and top diffractogram
- P42-VI-C second diffractogram
- P42- VI-D third diffractogram
- P42-VI-E fourth diffractogram
- P42-VII fifth diffractogram
- P42-VIII sixthth diffractogram
- P42-IX sixth and bottom diffractogram
- Fig. 8 shows the dissolution profile of 25mg Viagra tablets in 0.01M hydrochloric acid.
- Fig. 9 shows the dissolution profile of 50mg Viagra tablets in 0.01M hydrochloric acid.
- Fig. 10 shows the dissolution profile of lOOmg Viagra tablets in 0.01M hydrochloric acid.
- Fig. 11 shows the dissolution profile in 0.01M hydrochloric acid of the sustained-release core development formulation no. 16CF25/026 comprising sildenafil citrate, as described in Example 67.
- Fig. 12 shows the dissolution profile in 0.01M hydrochloric acid of the immediate - release development formulation no. 16CF25/027 comprising sildenafil citrate, as described in Example 67.
- Fig. 13 shows the dissolution profile of 25mg Viagra tablets in pH 4.5 phosphate buffer.
- Fig. 14 shows the dissolution profile of 50mg Viagra tablets in pH 4.5 phosphate buffer.
- Fig. 15 shows the dissolution profile of lOOmg Viagra tablets in pH 4.5 phosphate buffer.
- Fig. 16 shows the dissolution profile in pH 4.5 phosphate buffer of the sustained-release core development formulation no. 16CF25/026 comprising sildenafil citrate, as de- scribed in Example 67.
- Fig. 17 shows the dissolution profile in pH 4.5 phosphate buffer of the immediate -release development formulation no. 16CF25/027 comprising sildenafil citrate, as described in Example 67.
- Fig. 18 shows the dissolution profile of 25mg Viagra tablets in pH 6.8 phosphate buffer with 0.125% CTAB.
- Fig. 19 shows the dissolution profile of 50mg Viagra tablets in pH 6.8 phosphate buffer with 0.125% CTAB.
- Fig. 20 shows the dissolution profile of lOOmg Viagra tablets in in pH 6.8 phosphate buffer with 0.125% CTAB.
- Fig. 21 shows the dissolution profile in pH 6.8 phosphate buffer with 0.125% CTAB of the sustained-release core development formulation no. 16CF25/026 comprising sildenafil citrate, as described in Example 67.
- Fig. 22 shows the dissolution profile in pH 6.8 phosphate buffer with 0.125% CTAB of the immediate -re lease development formulation no. 16CF25/027 comprising sildenafil citrate, as described in Example 67.
- Fig. 23 shows the dissolution profile in 0.01M hydrochloric acid of the immediate - release development formulation no. 16CF25/033 comprising sildenafil free base, as described in Example 68.
- Fig. 24 shows the dissolution profile in pH 4.5 phosphate buffer of the immediate -release development formulation no. 16CF25/033 comprising sildenafil free base, as described in Example 68.
- alkyl refers to a linear or branched chain saturated hydrocarbon radical.
- a "C n - m alkyl” refers to an alkyl having from n to m carbon atoms.
- an alkyl group may be a Ci-25 alkyl group, a Ci-24 alkyl group, a Ce-24 alkyl group, a Ci-10 alkyl group, a Ci-6 alkyl group or a C1-4 alkyl group.
- Examples of a Ci-10 alkyl group are methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl or decyl.
- Ci-6 alkyl groups are methyl, ethyl, propyl, butyl, pentyl or hexyl.
- C1-4 alkyl groups are methyl, ethyl, i-propyl, n-propyl, t-butyl, s-butyl or n-butyl. If the term "alkyl" is used without a prefix specifying the number of carbons anywhere herein, it has from 1 to 4 carbons.
- alkenyl refers to a linear or branched chain hydrocarbon radical comprising one or more double bonds.
- a "C n - m alkenyl” refers to an alkenyl having from n to m carbon atoms.
- an alkenyl group may be a C2-25 alkenyl group, a C2-24 alkenyl group, a Ce-24 alkenyl group, a C2-10 alkenyl group, a C2-6 alkenyl group or a C2-4 alkenyl group.
- Examples of a C2-10 alkenyl group are ethenyl (vinyl), propenyl, butenyl, pen- tenyl, hexenyl, heptenyl, octenyl, nonenyl or decenyl.
- Examples of C2-6 alkenyl groups are ethenyl, propenyl, butenyl, pentenyl or hexenyl.
- Examples of C2-4 alkenyl groups are ethenyl, i-propenyl, n-propenyl, s-butenyl or n-butenyl.
- Alkenyl groups typically comprise one or two double bonds.
- co-crystal (or "co crystal” or “cocrystal”) as used herein means a solid that is a crystalline single phase material comprising two or more different molecular and/or ionic compounds which are neither solvents nor simple salts (S. Aitipamula et al. "Poly- morphs, Salts, and Cocrystals: What's in a Name?", Cryst. Growth Des., 2012, 12 (5), pp
- the two or more different molecular and/or ionic compounds in a co-crystal are generally compounds which are themselves solid at room temperature (i.e. solids at 22 °C). They are typically present in the co-crystal in a definite stoichiometric ratio.
- one of the two or more different molecular and/or ionic compounds is an API, sildenafil, and another of the two or more different molecular and/or ionic compounds is a co-crystal former.
- the co-crystals of the present invention are pharmaceu- tical co-crystals.
- a pharmaceutical co-crystal is a crystalline single phase material comprising an API and one or more unique co-crystal formers, typically in a stoichiometric ratio.
- Each of the one or more co-crystal formers is a molecular or an ionic compound that is a solid at room temperature. Solvates (including hydrates) of an API that do not further comprise a co-crystal former are therefore not considered to be co-crystals.
- a pharmaceutical co-crystal may how- ever include one or more solvent (e.g. acetonitrile, or water) molecules in the crystal lattice which comprises the API and the one or more unique co-crystal formers.
- Co-crystals can be constructed through several types of interaction, including hydrogen bonding (H -bonding), pi stacking, and van der Waals forces.
- H -bonding hydrogen bonding
- pi stacking pi stacking
- van der Waals forces van der Waals forces
- co-crystals often rely on hydrogen-bonded assemblies between neutral molecules of an API and another (co-crystal former) component.
- the dosage forms of the present invention are proven to work as pde5 inhibitors and find use in the treatment of male erectile dysfunction.
- Fresh frozen human penis was obtained from HAM (Pennsylvania). Tissue was thawed at room temperature, the corpus cavernosum was dissected from the penis to yield approximately 2-4 g of tissue and the following isolation protocol was followed. Tissue was coarsely chopped in ice-cold isotonic buffer (35 ml) con- taining 250 mm sucrose, 1 mM EDTA, 0.5 mM PMSF and 20 mM HEPES, pH 7.2, and the mixture subjected to brief (1 min.) treatment with a Silversen mixer/emulsifier.
- Homogenates were prepared using homogeniser tubes with Teflon pestles and a soluble fraction was prepared by centrifugation at 100,000 x g for 60 min. at 4°C. 10 ml of high speed supernatant was applied to a Pharmacia Mono Q anion exchange column (1 ml bed volume) equilibrated with buffer containing 1 mM EDTA, 0.5 mM PMSF and 20 mM HEPES, pH 7.2 (chromatography buffer). The column was then washed with 5 bed volumes of chromatography buffer, after which PDEs were eluted using a continuous gradient of 0-500 mM NaCl (total volume 35 ml) and 1 ml fractions collected.
- Fraction II hydro lysed cGMP and cAMP, with the latter activity being stimulated in the presence of cGMP, and was classified as PDEn, whilst fraction III was cAMP selective and this activity was inhibited in the presence of cGMP, consistent with PDEm activity.
- the above investigation identified three PDE isoenzymes in human corpus cavernosum tissue.
- the predominant PDE was the cGMP-specific PDEv, whilst cGMP- stimulated cAMP PDE n and cGMP-inhibited cAMP PDE m were also present.
- the formulations of the present inventions are tested in vitro and found to be potent and selective inhibitors of the cGMP-specific PDEv.
- relaxation of the corpus cavernosum tissue and consequent penile erection is presumably mediated by elevation of cGMP levels in the said tissue, by virtue of the PDE inhibitory profile of the compounds of the inven- tion.
- the sildenafil summary shows the in vitro dissolution profiles for the two elements of the formulation, the inner sustained release core with 80mg sildenafil and the outer core which is an ODT with 20mg sildenafil.
- Guidance is to place the tablet under the tongue for 60 seconds (see caffeine summary where we tested how much caffeine dissolved by doing this as substitute for Viagra) then swallow the tablet. Described herein is our model of how the tablet dissolves based on different pH levels We assume 20% will go into blood plasma PK from the sublingual element (some will go normal route so even if 30% dissolves not all will go sublingual). This would give us 4mg sublingual which according to other papers could deliver 4 to 5 times the dose in systemic circulation than normal, i.e.
- sildenafil in plasma somewhere around to a normal dose of 100 mg with about 112% to 116% of AUC (so within 505b2 guidelines) modelling and or testing in humans will confirm.
- a two component tablet is preferred the sildenafil summary shows the in vitro dissolution profiles for the two elements of the formulation.
- the present invention relates to novel pharmaceutical formulations, in particular to modified release formulations for the delivery of a therapeutically useful amount of sildenafil.
- Sildenafil citrate (Viagra®) is currently approved for the treatment of erectile dysfunction in male humans.
- Sildenafil is generically described in US patent 5,250,534 as a selective cGMP PDE inhibitor useful in the treatment of cardiovascular disorders such as angina, hypertension, heart failure and atherosclerosis.
- Sildenafil is specifically described in US patent 6,204,383 Bl as an agent with pharmaceutical utility in the treatment of male sexual dysfunction (MED).
- nitric oxide in the corpus cavernosum during sexual stimulation.
- NO nitric oxide
- cGMP cyclic guanosine monophosphate
- Sildenafil has no direct relaxant effect on isolated human corpus cavernosum, but enhances the effect of nitric oxide by inhibiting phosphodiesterase type5 (PDE5), which is re- sponsible for degradation of cGMP in the corpus cavernosum.
- sildenafil When sexual stimulation causes local release of (NO), inhibition of PDE5 by sildenafil causes increased levels of cGMP in the corpus cavernosum, resulting in smooth muscle relaxation and inflow of blood to the corpus cavernosum. Sildenafil at recommended doses has no effect in the absence of sexual stimulation.
- Sildenafil citrate in currently marketed swallow tablets is available in three dosage forms i.e. 25 mg, 50 mg and 100 mg of sildenafil calculated as the free base.
- the time for maximum plasma concentrations of sildenafil is between 0.5 and 2 hours when the patient is in a fasting state, but is delayed, on average, by 1 hour when sildenafil is taken with a high fat meal. After the initial variable delay in the onset of action, a therapeutic effect is maintained for approximately 2 hours, followed by a diminished response for about a further 2 hours.
- sildenafil can result in the inhibition of other phosphodiesterases, for example PDE 2 and PDE 6.
- PDE2 has been linked to the control of cardiac contractility
- PDE6 is found in the retina and is involved in the phototransduction pathway of the retina.
- a common side-effect of sildenafil when delivered by currently marketed swallow tablets is "blue haze", i.e. a transient dose-related impairment of colour discrimination, which is consistent with PDE6 inhibition.
- Warnings are provided on package inserts for existing sildenafil products against administering sildenafil to patients with pre-existing cardiac conditions.
- Sildenafil free base i.e. 5-[2-ethoxy-5-(4-methylpiperazin-l -ylsulfonyl)phenyl]-l- methyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, is specifically described in US patent 6,204,383 Bl .
- Example 1 illustrates the preparation of solid sildenafil by the evaporation of a solution in a mixture of dichloromethane and methanol.
- Examples 2 and 6 describe a solution of sildenafil iodide in dichloromethane, but this is not isolated as a solid product.
- Sildenafil is currently marketed as the citrate salt in numerous countries including the
- Sildenafil citrate is the active ingredient in VIAGRA®, which is approved for use in the treatment of MED.
- VIAGRA® which is approved for use in the treatment of MED.
- the choice of an improved formulation of sildenafil to deliver a clinically optimal dose of sildenafil is not a matter of routine.
- Other PDE5 agents have recently become available on the market for the treatment of MED. These agents include: Cialis®, and Levitra®. These agents have a faster onset of action and longer duration when compared to Viagra®, thereby emphasising the current deficiencies of Viagra®.
- sexual dysfunction is greatly affected and indeed potentiated by stress and other factors including anxiety, uncertainty, time- restrictions, the need for a controlled or limited diet and a clinical environment.
- maximum benefit for the patient is achieved when therapy takes place in as natural an environment as possible, when there is no pressure to keep to a strict timetable as a result of a narrow time-window for therapeutic efficacy, and when there is freedom from distracting side-effects.
- the time interval between dosing and onset of therapeutic efficacy with currently marketed swallow tablets is between 0.5 and 2 hours for fasted healthy male volunteers, and is delayed by a median 60 minutes following a high fat meal. In the general population, this uncertainty is likely to be even greater, and represents a major disadvantage for a stress-free sexual environment. Hence there is a need for a formulation which provides a rapid and predictable onset of therapeutic effect.
- the sharp bolus of sildenafil released into the blood plasma by currently marketed tablets, as described in the art, is undesirable because of the risk of side-effects.
- the target population for therapy includes many people of above middle age who are potentially vulnerable to side-effects associated with cardiac contractility, and who may be tempted to increase the dose beyond the recommended level. Furthermore, even clinically non-significant side- effects such as temporary vision impairment represent unwanted distractions which may increase stress and anxiety.
- sildenafil formulations can be prepared which provide either a rapid and predictable onset of therapeutic efficacy, or a rapid and predictable onset of therapeutic efficacy combined with a prolonged duration of therapeutic efficacy and a control of excessive blood plasma levels and adverse events.
- Such formulations are less affected by fasting and are more suited for use in a relaxed environment which is appropriate to non-clinical sexual activity when compared with the conventional swallow tablets of the prior art.
- the present invention provides a modified release drug delivery dosage form which delivers sildenafil within the therapeutic window either more rapidly than the currently marketed swallow tablets containing sildenafil, or continuously for a long period of time compared to the currently marketed swallow tablets containing sildenafil, or more rap- idly and continuously for a long period of time when compared to currently marketed swallow tablets containing sildenafil.
- the modified release formulation of the present invention is a rapid release, or a delayed release, or a prolonged release formulation, or any combination of the aforementioned, optionally combined with a conventional release component.
- the therapeutic window for sildenafil may also be defined in terms of efficacy and side-effect profile.
- the lower threshold of the therapeutic window is equivalent to the threshold at which an adequate improvement in erectile function is delivered.
- the higher threshold of the therapeutic window is the threshold at which side-effects become unacceptable. It is apparent that the therapeutic window lies between the thresholds at which a substantially undiminished erectile response is achieved.
- the formulations of this invention are provided in a series of dosing strengths for the purposes of dose titration, i.e. they permit an individual patient to commence with a low dose formulation and increase the dose systematically until an optimal effect is achieved. It is a further feature of this invention that the formulations of this invention are designed to provide a release of sildenafil within the above-de- fined therapeutic window.
- sildenafil is to be understood to include sildenafil in the form of the free base and also in the form of a pharmaceutically acceptable salt.
- Sildenafil free base is the compound 5-[2-ethoxy-5-(4-methylpiperazin-l-ylsulfonyl) phenyll- methyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one which is the active moiety in ViagraTM.
- pharmaceutically acceptable salt thereof refers to salts which are physically, chemically and physiologically acceptable for either human or veterinary use. It should also be understood that sildenafil or a pharmaceutically acceptable salt thereof includes solutions, amorphous forms, and crystalline forms of sildenafil including solvates, hydrates, co-crystals and polymorphs.
- sildenafil should be released within the therapeutic window over a period of more than 2 and up to 24 hours, for example between 2 hours and 5 hours, preferably between 2 and 10 hours, and more preferably between 0.15 and 24 hours (i.e. from dose to dose).
- rapid release in the context of "modified release”, in the context of this specification, means a formulation by means of which a therapeutic blood plasma concentration of sildenafil is achieved more rapidly than with the swallow tablets of the prior art, particularly in the non-fasted state, and more particularly when taken with a high fat meal.
- delayed release in the context of "modified release”, in the context of this specification, is understood to indicate a formulation that is designed to retard the initial release of drug from the dosage form by a pre-determined interval of time.
- delayed release may be understood to mean retardation of release, when compared to the currently approved product which is described as releasing sildenafil between 0.15 and 2 hours when measured in the fasting state, and one hour longer when taken with a high fat meal.
- Prolonged release in the context of "modified release”, in the context of this specification, may be understood to indicate a formulation that is designed to maintain the release of drug over a period of time that is substantially greater than is achieved in the cur- rently marketed formulation.
- substantially greater means that the drug is released within the therapeutic window for longer than 2 hours with no marked diminution of therapeutic effect. Accordingly, sildenafil is released within the therapeutic window over a period of more than 2 and up to 24 hours, for example between 2 hours and 5 hours, preferably between 2 and 10 hours, and more preferably between 2 and 24 hours. Rapid release formulations may be achieved by several different methodologies, which may be used alone or in combination.
- rapid release may be achieved by a dosage form of sildenafil comprising a rapidly dispersing wafer containing sildenafil or a pharmaceutically acceptable salt thereof which is placed on the tongue and dissolves in the mouth, for example within the buccal fluids.
- the wafer is dispersed and/or dissolved over a period of about 1 to 60 sec- onds, preferably about 1 to 30 seconds, most preferably about 1 to 10 seconds.
- the wafer is made from a freeze-dried compact containing sildenafil or a pharmaceutically acceptable salt thereof, in a matrix of a buccal fluid-dispersible polymer such as gelatine and a polysaccharide such as mannitol.
- Sildenafil is dissolved or dispersed into a suspension of mannitol and gelatine prior to filling into blister cavities. These liquid filled blisters are then conveyed through a liquid nitrogen freezing tunnel for freezing and then into a freeze dryer where the solvent is removed leaving behind a highly porous wafer loaded with sildenafil. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61-72 (2000), M J Rathbone, J Hadgraft & M S Roberts in Modified Release Drug Delivery Sys- tems, Marcel Dekker, New York, 2003, US Patent No. 4,642,903 and US Patent No
- rapid release of sildenafil may be provided by the blending and compression of sildenafil with water soluble excipients, such as a sugar such as but not limited to mannitol, and an effervescence agent, at low compression forces.
- water soluble excipients such as a sugar such as but not limited to mannitol
- effervescence agent an effervescence agent
- the low compression forces lead to the formation of a highly porous tablet which disintegrates rapidly. Rapid disintegration is further aided by the inclusion of the effervescence agent, which in the context of this specification is defined as one or more agents which produce carbon dioxide upon contact with buccal, gastric, or intestinal fluids.
- effervescence is derived by the reaction which takes place between alkali metal carbonates or bicarbonates and organic acids such as citric acid or tartaric acid to release carbon dioxide. Effervescence may also result from the inclusion of a carbonate or bicarbonate alone to react with acidic gastrointestinal fluids.
- the porous tablet disperses over a period of about 1 to 60 seconds, preferably about 1 to 45 seconds, most preferably about 1 to 30 seconds.
- sildenafil rapid release of sildenafil may be achieved by blending and compressing sildenafil with a suitable sugar such as but not limited to sucrose which has been melt- spun to form a mass of thin filaments with a high surface area.
- a suitable sugar such as but not limited to sucrose which has been melt- spun to form a mass of thin filaments with a high surface area.
- the resulting tablets are highly porous. Upon contact with buccal fluids, they disintegrate rapidly as the mass of thin filaments dissolves. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61-72 (2000) and US Patent No 4,855,326 which are incorporated herein by reference.
- rapid release of sildenafil may be achieved by blending and compressing sildenafil with a low mould ability saccharide (e.g. such as but not limited to lactose and mannitol) which has been granulated using a high mould ability saccharide (e.g. such as but not limited to maltose and maltitol) as a binder.
- a low mould ability saccharide e.g. such as but not limited to lactose and mannitol
- a high mould ability saccharide e.g. such as but not limited to maltose and maltitol
- the resulting tablets possess characteristics which enable them to dissolve rapidly on contact with aqueous fluids, typically within about 1 to 60 seconds, preferably about 1 to 30 seconds, most preferably about 1 to 15 seconds. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61 -72 (2000) and US Patent No
- rapid release of sildenafil may be achieved by blending and compress- ing sildenafil with a disintegrating agent (e.g. such as but not limited to carboxymethylcellu- lose) and a swelling agent (e.g. such as but not limited to modified starch, e.g. Sodium Starch Glycolate) to produce a rapidly disintegrable tablet which preferably on contact with aqueous fluids disperses over a period of about 1 to 90 seconds, preferably about 1 to 60 seconds, most preferably about 1 to 30 seconds.
- a disintegrating agent e.g. such as but not limited to carboxymethylcellu- lose
- a swelling agent e.g. such as but not limited to modified starch, e.g. Sodium Starch Glycolate
- a further embodiment of the present invention relates to the use of taste masking agents and flavours, or the use of forms of sildenafil, for example a suitable salt form, which have a pleasant or acceptable taste.
- One way of augmenting the rapid release achievable by a suitable choice of formulation is to utilise a salt of sildenafil which is very soluble in saliva or in gastric fluid.
- Yet another way of augmenting the rapid release achieved by a suitable choice of formulation is to utilise an amorphous form of a salt of sildenafil or sildenafil free base.
- the amorphous or crystalline form of a salt of sildenafil or sildenafil free base may be dispersed or adsorbed in a thin layer over a high surface area inert substrate.
- Suitable substrates include but are not limited to: Amberlite ® XAD-4, Amberlite ® XAD-7, Amberlite ® XAD- 16, AMBERSORB ® 348F, AMBERSORB ® 563, AMBERSORB ® 572, Activated carbon, Activated carbon Darco ®, Activated carbon Darco ® G-60, Activated carbon Darco ® KB, Activated carbon Darco ® KB-B, Activated carbon Norit ®, silica gel high purity grades with high pore volume, for example about 0.75 cc/g and average pore diameter 60A.
- a solution of sildenafil or a sildenafil salt may be prepared by dissolving the free base or a suitable salt thereof in a suitable solvent or by contacting stoichiometric quantities of the acid and base components of the salt in water or in a solvent such as but not limited to metha- nol, ethanol, or dichloromethane or a mixture thereof, for example at a concentration of 1 % to 20% by weight.
- the solution is mixed with the inert substrate as defined above, and the required product is isolated by a by either vacuum evaporation or by spray drying.
- the resulting product has sildenafil or a salt thereof dispersed or adsorbed in a thin layer over a high surface area inert substrate.
- the dissolution of sildenafil salts may also be enhanced by reducing the particle size and hence increasing the surface area, for example by such methods as jet milling, ball milling, controlled spray drying, and supercritical fluid precipitation.
- Jet or fluid energy milling involves the use of high pressure air jets to forcibly collide particles together, suitably within a hollow toroidal mill chamber.
- the high kinetic energy of the air causes particles to impact with other particles with sufficient energy for fracture to occur. This process is repeated until a desired particle size range is obtained.
- the product is suitably removed from the apparatus with a particle size classifier.
- Ball milling of sildenafil salts involves the use of a hollow cylinder rotated along a horizontal longitudinal axis. Inside the cylinder, grinding beads are loaded to a level of 30 to 50% of the chamber volume together with sildenafil salt powder feedstock. The size of the beads depends upon the material being processed and the size of the mill chamber. The chamber is rotated at a suitable velocity to allow the beads to cascade and grind the sildenafil drug particles to a finer and desired size.
- a feature of ball milling is that it can be conducted in the dry state, alternatively in a wet state in which sildenafil feedstock is supplied to the milling chamber as a suspension in a fluid. Drug particles of a fine and defined particle size can be manufactured by means of supercritical fluid precipitation.
- One suitable procedure is rapid expansion of a supercritical solution (RESS), in which sildenafil drug particles are formed as a result of the rapid expansion of a supercritical fluid containing dissolved sildenafil.
- a second procedure is gas anti-solvent recrystallisation (GAS) in which the supercritical fluid acts as an anti-solvent and causes sildenafil precipitation from a solution.
- GAS gas anti-solvent recrystallisation
- SEDS solution enhanced dispersion
- SEDS solution enhanced dispersion
- SEDS solution enhanced dispersion
- Multiple repeti- tions of these techniques is also one method for producing novel polymorphic forms which may have greater aqueous solubility and hence be more suitable for rapid release formulations.
- ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 5 microns. More preferably ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 3 microns. Even more preferably ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 1 micron. Most preferably ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 0.5 microns.
- Dispersants and/or other physical stabilisers may be added to prevent aggregation of ultrafine particles and the consequent reduction in the rate of dissolution.
- suitable dispersants and stabilisers and methods for their use can be found in Dissolution technology, L J Leeson and J T Carstensen (eds), APS, Washington 1974, and C.G. Liversedge et al., Int J Pharm Vol 125, pages 309-313, 1995), which publications are incorporated herein by reference.
- Useful stabilisers include polymers and surfactants e.g. hypromellose, hydroxypro- pylcellulose, mannitol, gelatine, tragacanth, acacia, sorbitan esters, glycerol monostearate, methylcellulose, carboxymethylcellulose, sodium lauryl sulphate, polyvinylpyrrolidone, poly- oxyethylene sorbitan fatty acid esters, polyoxyethylene alkly ethers, polyethylene glycols, block co-polymers of ethylene oxide and propylene oxide.
- surfactants e.g. hypromellose, hydroxypro- pylcellulose, mannitol, gelatine, tragacanth, acacia
- sorbitan esters glycerol monostearate, methylcellulose, carboxymethylcellulose, sodium lauryl sulphate, polyvinylpyrrolidone, poly- oxyethylene sorbitan fatty acid esters, poly
- Delayed release of sildenafil can be achieved by means of a physical barrier or coating which delays exposure of the active material to the buccal, gastric, or intestinal fluids.
- One technique which provides delayed release involves the application of a coating of a fluid resistant barrier to a single dosage unit, or to a multiparticulate dosage unit, for example one composed of beadlets, pellets, spheroids, minitablets and/or granules.
- These coatings can be designed to dissolve at a specific pH range, for example an enteric coating which dissolves at a pH greater than 5.0.
- Typical pH-dependent polymers suitable for coating dosage forms include the following:
- hydroxypropylmethylcellulose phthalate 50 which dissolves at about pH 4.8
- hydroxypropylmethylcellulose phthalate 55 which dissolves at about pH 5.2
- polyvinylacetate phthalate which dissolves at about pH 5.0
- methacrylic acid-methyl methacrylate copolymer (1 : 1) which dissolves at about pH 6.0 methacrylic acid-methyl methacrylate copolymer (2: 1), which dissolves at pH 6.5-7.5 methacrylic acid-ethyl acrylate copolymer (2: 1), which dissolves at about pH 5.5
- hydroxypropylmethylcellulose acetate succinate which dissolves at about pH 7.0
- poly(methylvinylether/maleic acid) monoethylester which dissolves at pH 4.5 -5.0
- poly(methylvinylether/maleic acid)n-butyl ester which dissolves at about pH 5.4
- non-pH-dependant coating may be used, which initially impedes the ingress of aqueous fluid, but subsequently erodes and/or dissolves to expose the active agent to dissolution.
- Typical non-pH-dependent polymers suitable for coating dosage forms (single or multiparticulate) to provide a fluid resistant barrier which subsequently erodes or dissolves include, but are not restricted to acacia, alginate, amylase, beeswax, carboxymethylcellulose, carnuba wax, cellulose acetate, cholesterol, ethylcellulose, fatty acids, gelatine, glyceryl be- henate, glyceryl monostearate, glyceryl monodistearate, glyceryl tripalmitate, hypromellose, hydroxypropylcellulose, hydrogenated vegetable oil, lecithin, methylcellulose, paraffin wax, pectin, polyethylene glycol, polycapro lactone, polyglycolic acid, polylactic acid, polygly
- Delayed release of sildenafil may also be achieved by a fluid resistant barrier which combines one or more pH-dependant polymers optionally with one or more non-pH-depend- ant polymers.
- delayed release dosage forms include enteric coated tablets or enteric coated multiparticulate formulations, in which drug-loaded multi-particulate spheres are coated with methacrylic acid-methyl methacrylate co-polymers such as Eudragit LI 00-55, Eudragit L30D-55, or Eudragit FS 30D or Eudragit S100/S12.5.
- methacrylic acid-methyl methacrylate co-polymers such as Eudragit LI 00-55, Eudragit L30D-55, or Eudragit FS 30D or Eudragit S100/S12.5.
- Such formulations will not release sildenafil in the acidic environment of the stomach but only on exposure to the higher pH typically found in the small and large intestine (pH range 5 to 8).
- An enteric coated tablet illustrating one aspect of this invention may be a single-layer tablet or a multi-layer tablet, such as a bi- or tri-layer tablet, wherein the active agent is present in one or more discrete lay- ers within the compressed tablet form.
- the discrete tablet layers can be arranged to provide modified or non-modified release of active agent.
- General descriptions and methods for the preparation of suitable tablets may be found in Aqueous polymeric coatings for pharmaceutical dosage forms, J W McGinty (ed), Marcel Dekker, 1989, New York, and in in Microencapsulation and related drug processes, P Deasy, Marcel Dekker, 1984, New York, which ref- cations are incorporated herein by reference.
- a capsule can be prepared in which the active dose is provided in the form of sildenafil beads and is divided into two or more parts, each part having a non-pH-depend- ant protective coat of different thickness, which takes a different time to erode.
- Suitable non- pH-dependent coating materials have already been described above. Further information can be found in J R Robinson & V H Lee (eds) in Controlled Drug Delivery, second edition, Marcel Dekker, New York, 1987 , V Ranade & M A Hollinger in Drug Delivery Systems, second edition, CRC Press, Boca Raton, 2004 and M J Rathbone, J Hadgraft & M S Roberts in Modified Release Drug Delivery Systems, Marcel Dekker, New York, 2003 which are incorporated herein by reference.
- Modified release may also be provided in the form of prolonged release.
- a prolonged release dosage form may consist of a matrix dosage unit, such as a hydrophilic and/or an erodible matrix, usually in tablet form. Release from such a unit can be controlled by a number of mechanisms, such as dissolution, erosion, diffusion, osmotic pressure or any combination thereof.
- Embodiment of prolonged release dosage forms may utilise excipients which control sildenafil release by more than one formal mechanism.
- An erosion controlled prolonged release dosage unit can be achieved by compressing sildenafil with a slowly dissolvable and/or erodable polymeric material into a tablet form. Release of sildenafil occurs as the polymer dissolves and/or erodes away.
- Suitable polymers include but are not restricted to glyceryl monostearate, acrylic resins, ethylcellulose, stearyl alcohol, hydroxypropylcellulose, carboxymethylcellulose, hypromellose, methylcellulose, hy- droxyethylmethylcellulose, sodium carboxymethylcellulose.
- a diffusion controlled prolonged release dosage form may be produced by compressing a water-swellable hydrophilic polymer in combination with sildenafil drug substance.
- Such systems are often referred to as "hydrophilic matrices" or “swellable-soluble” systems. Water continues to penetrate the matrix causing the swelling of the hydrophilic polymer. The gelatinous layer that is formed, retards the rate of ingress of water into the matrix and the flux of drug out of the matrix. Sildenafil is released from such matrices either by diffusion through the gel layer or by erosion and/or dissolution of the gel layer.
- Suitable materials would include any pharmaceutically acceptable excipient which can swell and form a gelatinous mass upon hydration, for example, hydroxypropylmethylcellulose, and xanthan gum. Further information and descriptions of such dosage forms can be found in Controlled Drug Delivery, sec- ond edition, J R Robinson & V H Lee (editors), Marcel Dekker, New York, 1987 which publication is incorporated herein by reference.
- An osmosis controlled prolonged release dosage form may be produced by compressing sildenafil in combination with an osmagent into a tablet matrix core formulation.
- This matrix core is then in part coated with a semi-permeable membrane in known manner, utilis- ing such polymers such as methacrylates, ethylcellulose, and cellulose acetate.
- Aqueous fluids are drawn by osmosis from the exterior environment across the membrane at a controlled rate into the core, causing dissolution of both sildenafil and the osmogent and increased pressure within the matrix core. The pressure forces the solubilised sildenafil out through a specially created aperture or passageway.
- osmagents include but are not restricted to sodium chloride, potassium chloride, lithium chloride, magnesium chloride, magnesium sulphate, lithium sulphate, sodium sulphate, potassium sulphate, citric acid, mannitol, ribose, arabinose, galactose, leucine, glycine, fructose, sucrose, sodium and other bicarbonates. Further information can be found in the scientific and patent literature, for example: Controlled Drug Delivery, second edition, J R Robinson & V H Lee (editors), Marcel Dekker, New York, 1987, Modified Release Drug Delivery Systems, M J Rathbone, J Hadgraft & M S
- Prolonged release can also be achieved by applying a porous or semipermeable membrane coat onto a tablet surface by the application of such polymers such as methacrylates, ethylcellulose, and cellulose acetate. Release from such systems can occur by more than one of the mechanisms described above, for example a combination of dissolution, diffusion, erosion, and osmosis.
- prolonged release can be achieved by coating multiparticulates with semipermeable membranes.
- the multiparticulates include drug-coated substrates, such as lactose beads, and drug-containing substrates, such as drug-containing lactose spheres.
- compositions of the present invention provide a single modified release formulation of sildenafil or a combination of two or more taken from the various modified and unmodified release forms of sildenafil: rapid release, conventional release, pulsed release, delayed release, and prolonged release. Therefore the compositions of this invention have a component or a combination of components which possess one of the following release characteristics:
- One preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with duration of activity of approximately 2 hours.
- Such a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conventionally be necessary.
- Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, and who wishes to avoid distracting or harmful side-effects.
- Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided with an initial delay of 0.15 to 2 hours, and with duration of activity of approximately 4 hours.
- Such a formulation may comprise a conventional release component at a lower dose than would conventionally be necessary, combined with a delayed release component.
- Such a formulation is useful to a patient who expects a sexual encounter at a time which is not less than 2 hours in the future, but does not wish to be restricted by recent or imminent food intake, and who wishes to avoid distracting or harmful side-effect
- Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with duration of activity of approximately 4 hours.
- a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conventionally be necessary, and a delayed release component.
- Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, but who is uncertain of the exact timing, and who wishes to avoid distracting or harmful side-effects.
- Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided with an initial delay of 0.15 to 2 hours, and with a duration of activity of approximately 12 hours.
- a formulation may comprise a conventional release component at a lower dose than would conventionally be necessary com- bined with either a prolonged release component or a pulsed release component.
- such a formulation is useful to a patient who expects a sexual encounter at a time which is not less than 2 hours in the future, but is very uncertain of the timing and who does not wish to be restricted by recent or imminent food intake, and who wishes to avoid distracting or harmful side-effects.
- a b.i.d. presentation of this formulation may be employed for the purposes of continu- ous treatment.
- Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with a duration of activity of approximately 12 hours.
- a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conventionally be necessary, and a prolonged release component.
- Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, and who wishes to be prepared for sexual activity for an extended period of time, and who wishes to avoid distracting or harmful side-effects.
- Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided with an initial delay of 0.15 to 2 hours, and with duration of activity of approximately 16-24 hours.
- a formulation may comprise a conventional release component at a lower dose than would conventionally be necessary combined with either a prolonged release component.
- Such a formulation is useful to a patient who expects a sexual encounter at a time which is not less than 2 hours in the future, but is very uncertain of the timing and who wishes to be prepared for sexual activity for an extended period of time, and who does not wish to be restricted by recent or imminent food intake, and who wishes to avoid distracting or harmful side-effects.
- Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with a duration of activity of approximately 16-24 hours.
- a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conven- tionally be necessary, and a prolonged release component.
- Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, and who wishes to be prepared for sexual activity for a much extended period of time, and who wishes to avoid distracting or harmful side-effects. It should be appreciated that this approach to providing a variety of specially designed formulations to address specific circumstances is es- pecially useful for over-the-counter products where the patient is able to self-diagnose and choose the most appropriate product for his specific needs.
- these formulations may provide effective therapy for female patients suffering from sexual disorders including but not limited to lack of clitoral arousal. This is especially surprising since the innovator company have reported lack of effi- cacy for sildenafil in female patients.
- the dose of sildenafil calculated as the free base, will be adjusted in consideration of the amount of sildenafil in each component and the total amount so as to ensure that blood plasma levels remain within the therapeutic window whilst achieving the effect desired and described above.
- the quantity of sildenafil required in each component of each formulation can be determined by the skilled worker from the information provided in this invention. Firstly the target pharmacokinetic profile for the formulation is selected in line with the objects of the present invention. Then, from knowledge of the therapeutic window as defined herein, the mean rate of elimination of sildenafil in the body, and the release profile of sildenafil from each component, it is a matter of routine experimentation to establish the necessary quantity of sildenafil in each component.
- each of the formulations of this invention will be provided in a range of strengths to permit titration of dose for individual patients.
- the compositions are in unit dosage form.
- Unit dosage forms for oral administration may be in tablet or capsule form and may as necessary contain conventional ex- cipients such as binding agents, fillers, lubricants, glidants, disintegrants, effervescent agents, and wetting agents.
- binding agents include but are not limited to: acacia, alginic acid, car- boxymethylcellulose calcium, carboxymethylcellulose sodium, dextrin, dextrose, ethylcellu- lose, gelatin, liquid glucose, guar gum, hydroxyethyl cellulose, hydroxypropyl cellulose, hy- droxypropyl methylcellulose, magnesium aluminium silicate, maltodextrin, methyl cellulose, polymethacrylates, polyvinylpyrrolidone, pregelatinised starch, sodium alginate, sorbitol, starch, syrup, tragacanth.
- fillers include but are not limited to: calcium carbonate, calcium phosphate, calcium sulphate, carboxymethylcellulose calcium, carboxymethylcellulose sodium, compressible sugar, confectioner's sugar, dextrates, dextrin, dextrose, dibasic calcium phosphate dihydrate, dibasic calcium phosphate, fructose, glyceryl palmitostearate, glycine, hydro- genated vegetable oil-type 1 , kaolin, lactose, maize starch, magnesium carbonate, magnesium oxide, maltodextrin, mannitol, microcrystalline cellulose, polymethacrylates, potassium chloride, powdered cellulose, pregelatinised starch, sodium chloride, sorbitol, starch, sucrose, sugar spheres, talc, tribasic calcium phosphate, xylitol.
- lubricants include but are not limited to: calcium stearate, glyceryl monostearate, glyceryl palmitostearate, magnesium stearate, microcrystalline cellulose, sodium benzoate, sodium chloride, sodium lauryl sulphate, stearic acid, sodium stearyl fumarate, talc, zinc stearate.
- glidants include but are not limited to: colloidal silicon dioxide, powdered cellulose, magnesium trisilicate, silicon dioxide, talc.
- disintegrants include but are not limited to: alginic acid, carboxymethylcellulose calcium, carboxymethylcellulose sodium, colloidal silicon dioxide, croscarmellose sodium, crospovidone, guar gum, magnesium aluminium silicate, microcrystalline cellulose, methyl cellulose, polyvinylpyrrolidone, polacrilin potassium, pregelatinised starch, sodium alginate, sodium lauryl sulphate, sodium starch glycolate.
- effervescent agents are effervescent couples such as an organic acid and a metal carbonate or bicarbonate.
- Suitable organic acids include but are not limited: citric acid, tartaric acid, malic acid, fumaric acid, adipic acid, succinic acid, and alginic acid, and anhydrides and acid salts.
- Suitable carbonates and bicarbonates include, for example, sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate, magnesium car- bonate, sodium glycine carbonate, L-lysine carbonate and arginine carbonate.
- only the base component of the effervescent couple may be present.
- the solid oral compositions may be prepared by conventional methods of blending, filling or tableting.
- the tablets may be coated according to methods known in normal phar- maceutical practice. For example see Pharmaceutical dosage forms: tablets, Volume 1 second edition, H A Lieberman, L Lachman and J B Schwartz (eds) Marcel Dekkker, 1989, New York and G C Cole & J Hogan in Pharmaceutical coating technology, Taylor & Francis, London, 1995 which are herein included by reference.
- Formulations of the present invention may be used for the treatment of human male erectile dysfunction, which method comprises administering a formulation of the present invention comprising an effective amount of sildenafil and/or a pharmaceutically acceptable salt thereof to a sufferer in need thereof.
- Formulations of the present invention may also be used for the treatment of human female sexual dysfunction, which method comprises administering a formulation of the pre- sent invention comprising an effective amount of sildenafil and/or a pharmaceutically acceptable salt thereof to a sufferer in need thereof.
- Formulations of the present invention may also be used in the preparation of a medicament for use in the treatment of human male erectile dysfunction.
- Formulations of the present invention may also be used in the preparation of a medic- ament for use in the treatment of human female sexual dysfunction.
- the present invention also provides a pharmaceutical composition for use in the treatment of human male erectile dysfunction or human female sexual dysfunction in which the pharmaceutical composition is as defined in the present invention.
- Examples 52 to 65 hereinbelow it should be appreciated that the amount of active ingredient is not specified precisely but is readily calculated from a consideration of the target product profile required within the scope of the present invention without undue experimentation.
- the sildenafil citrate used in these examples may be replaced by other salts of sildenafil with compatible solubility properties.
- the present invention also relates to novel compounds and to their use in medical therapy, in particular to their use in the treatment and/or prophylaxis of disorders associated with PDE5 inhibition.
- the present invention also relates to processes for preparing these novel compounds.
- Sildenafil is generically described in US patent 5,250,534 as a selective cGMP PDE inhibitor useful in the treatment of cardiovascular disorders such as angina, hypertension, heart failure and atherosclerosis.
- Sildenafil free base i.e. 5-[2-ethoxy-5-(4-methylpiperazin-l -ylsulfonyl)phenyl]-l- methyl-3- propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, is specifically described in US patent 6,204,383 Bl as an agent with pharmaceutical utility in the treatment of male sexual dysfunction.
- Sildenafil citrate is the active ingredient in VIAGRA®, which is approved for use in the treatment of MED.
- the choice of a form of a pharmaceutical agent having a basic functional group is not a matter of routine.
- Pfizer the originator company
- markets the citrate salt could lead one skilled in the art to believe that other forms were less preferred since the citrate salt is an unusual choice of salt to market.
- the choice of salts of sildenafil other than the prior art citrate salt is not therefore prima facie obvious in view of this technical prejudice and other concerns about the formation and properties of such salts.
- sildenafil other than the prior art citrate which are pharmaceutically acceptable. Such forms apart from finding use in medical therapy and as useful intermediates are also useful in providing new active ingredients containing the active moiety sildenafil which could form the basis for providing new value-added line extenders in the form of advantageous formulations or new uses.
- salts of sildenafil with certain long chain fatty acids can be prepared and have a pleasant and fully acceptable flavour which permits the use of formulations such as chewable tablets, chewing gum, and oral suspensions.
- the invention accordingly en- compasses such chewable tablets, chewing gum, and oral suspensions, but also is of value in swallow tablets and other conventional formulations. Accordingly, the present invention provides sildenafil salts with long chain fatty acids.
- the yield is established by comparison of the weights of cost-critical starting material and product making allowance for molecular weights and purities. Purities are established by hplc, gc or other conventional analytical method by means of a validated procedure and comparison with a reference standard. See for example Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA, ISBN 0-683-306472, page 597.
- Colour may be defined as the perception or subjective response of an observer to the objective stimulus of radiant energy in the visible spectrum extending over a range 400nm to 700nm in wavelength.
- hue or the quality by which one colour family is distinguished from another, such as red, yellow, blue, green and intermediate terms
- value or the quality that distinguishes a light colour from a dark one
- chroma or the quality that distinguishes a strong colour from a weak one, or the extent to which a colour differs from a grey of the same value.
- the perception of colour and colour matches is dependent on conditions of viewing and illumination. Determinations should be made using diffuse, uniform illumination under conditions that reduce shad- ows and nonspectral reflectance to a minimum.
- the surfaces of powders should be smoothed under gentle pressure so that a planar surface free from irregularities is presented.
- Liquids should be compared in matched colour-comparison tubes, against a white background. If results are found to vary with illumination, those obtained in natural or artificial daylight are to be considered correct. Colours of standards should be as close as possible to those of the test specimens for quantifying colour differences. Instrumental methods for measurement of colour provide more objective data than the subjective viewing of colours by a small number of individuals.
- the European Pharmacopoeia describes definitions and methods by which bulk density of a powder may be measured. Apart from the inherent density of a material which depends on factors such as crystal structure which is unpredictable, there is also the contribution of interp articulate void volume which is equally unpredictable.
- the bulk density is determined by measuring the volume of a known mass of powder, which has been passed through a screen, into a graduated cylinder. The tapped density is achieved by mechanically tapping a measuring cylinder containing a powder sample. After observing the initial volume, the cylinder is mechanically tapped, and volume readings are taken until little further volume change is observed.
- the ability of a powder to flow efficiently through manufacturing apparatus is a sig- nificant factor affecting the economics of manufacture and will vary according to the form of material. For example different salts, polymorphs, and pseudopolymorphs will have different inherent flow properties, though isolation procedures will also have an effect. Suitable definitions and methods of measurement are described in standard reference texts, for example European Pharmacopoeia 4 th edition 2001, and United States Pharmacopeia 24 rd edition 1999- 2003 and Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA. Measurement may be for example in terms of the angle of repose, which may be determined experimentally by a number of methods with slightly differing results. A typical method is to pour the powder in a conical heap on a level, flat surface and measure the included angle with the horizontal.
- Typical measurement procedure include the "Burning Rate Test” or "Fire Train Test” as defined in United States Department of Transportation and United Nations regulations (49 CFR 173 Appendix E and UN Recommendations on the Transport of Dangerous Goods, also EEC Directive 79/831 Annex Part A: Methods for the Determination of Physico-Chemical Properties 3.10 Flammability of Solids.
- a typical test involves applying a source of ignition to a powder "train” measuring 250 mm x 20 mm x 10 mm and measuring the rate at which the powder burns.
- Standard test methods are described in the major pharmacopoeias and are also referenced on the US Food and Drug Administation Web site.
- accelerated storage tests are performed by stor- age for a period of 1 year or more at elevated temperature (e.g.40°C) and at standard humidity conditions (e.g. 75% RH), with samples being taken at regular intervals of approximately 1 month and assayed for overall purity, specific impurities, and a general impurity screen.
- melting points and glass transition temperatures will differ greatly for different salts, polymorphs, pseudopoly- morphs, or other forms of a drug and are in essence unpredictable.
- Methods for measuring melting points are well-described in the European Pharmacopoeia 4 th edition 2001, and United States Pharmacopeia 24 rd edition 1999-2003.
- Various methods are acceptable but differ in detail, for example the melting point determined by the capillary method is the temperature at which the last solid particle of a compact column of a substance in a tube passes into the liquid phase.
- Suitable apparatus is described in the above mentioned publications and may be calibrated using melting point reference substances such as those of the World Health Organisation or other appropriate substances.
- Some materials have a tendency to change their physical form during storage, which can be a disadvantage in pharmaceutical manufacture. For example materials can settle and compact and lose their ability to low freely.
- One polymorphic form may wholly or partially convert to another over an uncertain time-frame, or solvates and hydrates may lose their solvent or water, and the resultant change in the physical properties of the drug substance can lead to a formulation with uncertain, unreliable, and unpredictable characteristics.
- Clearly a polymorphic conversion can only occur from a less stable to a more stable form, so there are advantages associated with thermodynamic stability, and the relative stability of a novel form is a priori unpredictable.
- hygroscopicity describes both the rate and the extent of water uptake. It is well established that hygroscopic products are difficult to handle and hence more expensive to formulate. Hygroscopicity is not a priori predictable, and an alternative salt may well be advantageous in this respect.
- Apparatus for measurement of moisture contents of samples under controlled humidity conditions is available commercially, e.g. from I Holland Ltd., Nottingham, U.K. Simple measurements may be made by monitoring the appearance and weight of samples exposed to atmospheres of known constant humidity and temperature, as described in, for example, The Merck Index 12 th edition, Merck and Co Inc.
- wetting is the ability of liquids to form boundary surfaces with solid materials, and is determined by measuring the contact angle which a liquid forms in contact with a solid. The smaller the contact angle the larger the wetting tendency. Wetting phenomena are described in Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA on pages 278-9. In order for immersion of a solid to occur, the liquid must displace air and spread over the surface of the solid.
- compositions / delivery range of technologies are very important and will differ in a non-predictable manner depending on the specific properties of the drug form.
- Pharmaceutical excipients are substances, other than the active pharmaceutical ingredient, that are used in the finished dosage form. There are very many widely differing excipients each with particular characteristics which form the basis of many widely differing formulations. Excipients and their properties are described in detail in the pharmaceutical literature, for example in Re- mington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA.
- the present invention provides amorphous, crystalline or liquid sildenafil salts with long chain fatty acids.
- the novel salts of sildenafil with long chain fatty acids may be in the form of an amorphous solid.
- amorphous sildenafil salts with long chain fatty acids may be used as an ingredient for inclusion in a range of formulations such as conventional tablets and capsules, in particular, a chewable tablet or formulated into a chewing gum or suspension or liquid for oral administration, or may be prepared in a form in which the salt is absorbed in a carrier, for example an excipient or a mixture of excipients for tabletting or other formulation, or as a solution in a wax or similar pharmaceutically acceptable polymer, such as PEG or PVA.
- the salt is formulated as a component of a sweet or chocolate based confection.
- sildenafil salts with long chain fatty acids may be in crystalline form. More than one crystalline form may be possible and such polymorphs and pseudo polymorphs including hydrates and solvates also form an aspect of this invention.
- sildenafil salts with long chain fatty acids may be in liquid form.
- Such liquids may be prepared by conventional methods such as dissolving a crystalline or amorphous material in a suitable solvent.
- Sildenafil is the active moiety in sildenafil citrate which is the active ingredient in VI ⁇
- fatty acid is understood by one skilled in the art and comprises a monobasic carboxylic acid with a carbon and hydrogen containing substituent group.
- Long chain is to be understood as comprising a substituent group with six or more carbon atoms.
- the carbon and hydrogen containing substituent group is a C6-C24 alkyl group or a C6-C24 alkenyl group, i.e. the long chain fatty acid is of formula RC(0)OH wherein R is C6-C24 alkyl or C6- C24 alkenyl.
- Examples of salted versions of sildenafil with long chain fatty acids include:
- Amorphous sildenafil heneicosanoic acid salt Amorphous sildenafil heptadecanoic acid salt
- sildenafil free base is described in US patent 6,204,383 Bl .
- a solution of sildenafil free base may be prepared from the commer- cially available citrate salt by basifying a suspension of the citrate salt in a mixture of water and dichloromethane to a pH of between 8 and 10 and separating the organic phase.
- Sildenafil base is obtained by evaporation of the organic phase.
- the long-chain fatty acids of this invention are available commercially from chemical suppliers such as Aldrich Chemical Company in the UK.
- a solution of a salt of sildenafil with a long-chain fatty acid may be prepared by contacting stoichiometric quantities of the acid and base components of the salt, for example by heating an equivalent of sildenafil base and the long chain fatty acid in hot absolute ethanol or methanol at a concentration of 1% to 20% by weight.
- a solution in dichloro- methane may be prepared by heating an equivalent of sildenafil base and the long chain fatty acid in dichloromethane at a similar range of concentrations.
- a mixture of these solvents or other solvents such as may be used.
- a miscible co-solvent may be used at a proportion of between 1 :10 and 10: 1.
- Suitable acids include decanoic acid, docosanoic acid, eicosanoic acid, heneicosanoic acid, heptadecanoic acid, lauric acid, myristic acid, non- adecanoic acid, nonanoic acid, octanoic acid, palmitic acid, pentadecanoic acid, stearic acid, tetracosanoic acid, tricosanoic acid, tridecanoic acid, undecanoic acid, undecylenic acid.
- Suitable co-solvents include n-propanol, propan-2-ol, acetone, 2-butanone, diethyl ether, toluene, and acetonitrile or a mixture thereof.
- Amorphous sildenafil long chain fatty acid salts are then isolated by either rapid vacuum evaporation, or spray drying.
- a concentration of between 2% and 40% weight/volume salt in solution, optionally at elevated temperature, may be used though a concentration of between 10 and 25% is preferred.
- Aqueous mixtures containing organic solvents may be spray dried in a closed loop spray dryer. If a closed loop spray drier is used the organic solvent content may be raised further up to 100%.
- the apparatus parameters are ad- justed to give an acceptable product by routine means, but control of outlet gas temperature and solvent content of the outlet gas is particularly important.
- the outlet temperature is kept above 40°C but below 70°C, more preferably below 50°C, and the solvent content of the outlet gases is kept below 2 grammes per 100 grammes, more preferably below 1.2 grammes per 100 grammes.
- spray drying is described in the following publications which are incorporated herein by reference: Spray-drying handbook 5th edition by K Masters, Longman Scientific & Technical, 1991 ISBN 0582062667; Spray- drying of pharmaceuticals for controlled release pulmonary drug delivery by Noha Patel, University of Bath, 2000; Spray-drying of particles intended for inhalation by Kristina Mos'em, Department of Pharmaceutics, Copenhagen, Danish University of Pharmaceutical Sciences 2003, ISBN 8778345243.
- the solution of sildenafil salt with a long chain fatty acid may be mixed with a suspension of one or more finely powdered excipients or excipients in solution before spray drying, thus preparing in one step a platform formulation for tabletting or other preparation of a drug product.
- Known techniques may be employed to coat the particles with enteric or other known coatings for control of drug release after administration.
- Drying to the full extent that is desirable for a stable pharmaceutical product is not always practicable during efficient use of the isolation apparatus, particularly in the case of spray drying, so in all the above procedures, a final air or vacuum drying step may be necessary to reduce residual water and solvent to an acceptable level.
- a solution of a salt of sildenafil may also be prepared by passing a solution of a strong acid salt of sildenafil or sildenafil in a solvent in which the fatty acid sildenafil salt is soluble through a strong base ion-exchange column in which the column material is in the desired fatty acid salt form.
- ion exchange resins are generally described in the literature, for example: Ion Exchange Resins 2nd edition by Robert Kunin, John Wiley and Sons, Inc. New York, 1958; The use of ion-ex - change resins as potential bioadhesive drug delivery systems, by Sarah J Jackson, University of Nottingham, UK 1999; Ion exchange resins by Robert Kunin, R.E.
- Suitable ion exchange resins include: AMBERJETTM 4200(C1), Amberlite ® IRA-400 (CI), Amberlite ® IRA-410, Amberlite ® IRA-900, Amberlite ® IRA-743, Dowex ® 1X2-100, Dowex ® 1X2- 200, Dowex ® 1X2-400, Dowex ® 1X4-50, Dowex ® 1X4-100, Dowex ® 1X4-200, Dowex ® 1X4-400, Dowex ® 1X8-50, Dowex ® 1X8-100, Dowex ® 1X8-200, Dowex ® 1X8-400, Dowex ® 21K CI, Dowex ® 2X8-100, Dowex ® 2X8-200, Dowex ® 2X8-400, Dowex ® 22 CI, Dowex ® MARATHON ® A, Dowex ® MARATHON ® A2, Dowex ® MSA-1, Dowex ® MSA-2, Dowex
- Crystalline salts of sildenafil with long chain fatty acids are prepared by trituration of the amorphous salts with one of the solvents (or a mixture of solvents) preferably from the following list of preferred solvents, i.e., diethyl ether, di-isopropyl ether, teri-butyl- methylether, di-n-butyl ether, butylvinylether, teri-butylvinylether, tetrahydrofuran, 1,4-diox- ane, n-heptane, n-hexane, cyclohexane, cyclopentane, toluene, o-xylene, m-xylene, p-xylene, dichloromethane, chloroform, carbon tetrachloride, chlorodibromofluoromethane.
- solvents i.e., diethyl ether, di-isopropy
- a solvent (or a mixture of solvents) from the following list may be used to triturate and crystallise amorphous sildenafil salts with long chain fatty acids: methyl ace- tate, ethyl acetate, propyl acetate, ethyl formate, isobutyl acetate, isobutyl formate, acetoni- trile, isobutyronitrile, acetone, butanone, isopropylmethylketone, isobutylmethylketone, tert- butylmethylketone, sec-butylmethylketone, w-butylmethylketone, cyclopentanone, cyclohexa- none.
- More polar solvents which also have utility for trituration are the following: water, methanol, ethanol, n-propanol, propan-2-ol, 1-butanol, isobutyl alcohol, cyclopentanol, 2-ethoxyethanol, 2-methyl-2-butanol, ethyleneglycol, teri-butanol, acetic acid, propionic acid.
- the solvents below may also be used for trituration: N,N-dime- thylformamide, ⁇ , ⁇ '-dimethylacetamide, N-methylpyrrolidone, formamide, anisole, sul- folane, nitromethane, ⁇ , ⁇ '-dimethylpropyleneurea, dimethylsulfoxide, benzene , chloroben- zene, 1 ,2-dichlorobenzene, 4-methylmorpholine, ⁇ , ⁇ , ⁇ ', ⁇ '-tetramethylethylenediamine, pyridine, diisopropylamine, triethylamine, aqueous ammonia.
- Suitable trituration techniques include addition of a small amount of solvent to the amorphous salt, insufficient to cause complete solution, and scratching with a glass rod and/or ultrasonication. Scratching is carried out in a glass tube for periods of approximately 5-10 minutes at a time, then leaving the tube to stand for 1 to 2 hours. Preferably the suspension/paste is brought onto the sides of the vessel during scratching to allow partial evaporation of the solvent. The procedure is then repeated several times. Alternatively the process may be automated by agitating the suspension/paste very vigorously with a small magnetic stirrer in the presence of some quartz anti-bumping granules or other abrasive material such as carborundum. Alternatively the mixture may be subjected to thermal shock by repeated cyclic ultra-freezing with liquid nitrogen followed by heating. Combinations of these techniques may be used.
- Another technique which may be employed is to raise the temperature of the amor- phous salt above the glass point and to raise and lower the temperature in a cyclic manner. This technique may be used in combination with thermal shock treatment and scratching.
- the crystalline product is dried under moderate vacuum to remove excess solvent but not such vigorous conditions as to desolvate any solvate that may have been formed.
- the existence of a crystalline salt may be demonstrated by a combination of techniques. Solution nmr and or elemental analysis is used to demonstrate that both acid and base components are present.
- the salt is characterised by one or more of the following techniques: infra-red spectroscopy, Raman spectroscopy, X-ray powder or single crystal diffraction, solid- state nmr, melting point, DSC, DTA, optical and electron microscopy.
- the salt may be pre- sent as a hydrate or a solvate.
- a crystalline salt has been prepared on the small scale by one of the techniques described above so that seeds are available and a suitable solvent has been identified
- other methods may be employed which are more suitable for large scale synthesis. For example, direct crystallisation from a solvent or solvent mixture which has been prepared by mixing the acid and base components optionally at elevated temperature. The solution is made supersaturated by cooling, partial evaporation, or addition of an antisolvent. Alternatively, crystallisa- tion from a supercritical fluid may be employed.
- a salt may be prepared by adding a soluble conjugate salt to a soluble salt of sildenafil in a solvent in which the target salt is insoluble, i.e.
- the salt of sildenafil with X is prepared by mixing sildenafil/Y with Z/X (a soluble salt of X).
- a salt of sildenafil with a long chain fatty acid may be prepared by adding an ammonium salt of a long chain fatty acid to a solution of a salt of sildenafil with a weak acid such as acetic acid or similar, preferably in the presence of seeds of the crystalline target salt.
- an antisolvent may be added (selected from those solvents which experimentally are found not to dissolve the salt, even with heating).
- non-crystalline sildenafil salts are used for these preparations since this minimises the presence of pre-existing crystal forms which may inhibit the production of alternative crystal- line products.
- hydrates and solvates may be treated with solvents or subjected to a range of different humidities to give other solvates or hydrates.
- the solvates or hydrates may be subjected to vacuum or oven drying/desolvation, or plunged into an immiscible hot solvent (for example xylene).
- crystalline forms may be subjected to a combination of very high pressure and optionally high temperature, for example melts, or amorphous material maintained above the glass temperature can be induced to crystallise to give a new product.
- More stable polymorphs may be generated by means of a "polymorph amplifier”. This procedure comprises the preparation of a stirred suspension of a crystalline salt in a selected solvent (approximately 5-10% in solution at the lower temperature) and raising the temperature until approximately 90-95% of the solid is dissolved, then allowing the suspension to cool slowly, with stirring, until most of the salt has crystallised. This procedure is repeated cyclically many times, and the product at each stage tested for any change in form. Crystallisation of novel crystal forms may be brought about by crystallising (for example by evaporation) on quartz or other active surfaces.
- the existence of polymorphs and pseudopolymorphs may be demonstrated by various techniques in combination.
- Solution NMR and/or elemental analysis is used to demonstrate that both acid and base components are present.
- the salt is characterised by one or more of the following techniques: infra-red spectroscopy, Raman spectroscopy, X-ray powder or sin- gle crystal diffraction, solid-state NMR, melting point, DSC, DTA, optical and electron microscopy, measurements of density, wetting angle, solubility, stability, and flow properties.
- fatty acid salts of sildenafil of this invention may also be prepared as solid or liquid solutions or dispersions in a liquid or polymeric carrier or matrix.
- Such matrix dispersions may be prepared in a variety of ways; the salt may be added to the matrix material either as a solid or in solution and the matrix material itself may also be either in the form of a solid (or liquid, as appropriate) or in solution. If both materials are solids then heating and stirring of a melt may be utilised to form a homogenous mixture before the product is cooled, and either ground to a powder, or left as a liquid or semi-liquid suitable for further formulation.
- Various techniques are known for the formation of suitable granules and platform products from melts, for example spray congealing techniques to produce pellets have been described by Kanig J.Pharm Sci 53, 188, 1964 and by Kreuschner et al. Acta Pharm. Tech. 23, 159, 1980.
- a liquid matrix may be used to dissolve the solid salt, or a solution of the matrix product may be formed by mixing a solution of the salt with a solution of the matrix material, and the solvent subsequently removed by evaporation or spray-drying.
- Suitable solvents for preparing solid or liquid solutions or dispersions include water, common alcohols, ketones, esters and, ethers.
- Particularly preferred solvents are water, methanol, etha- nol, n-propanol, propan-2-ol, 1-butanol, isobutyl alcohol, cyclopentanol, 2-ethoxyethanol, 2- methyl-2-butanol, ethyleneglycol, teri-butanol, acetone, butanone, isopropylmethylketone, isobutylmethylketone, teri-butylmethylketone, sec-butylmethylketone, ra-butylmethylketone, cyclopentanone, cyclohexanone, methyl acetate, ethyl acetate, propyl acetate, ethyl formate, isobutyl acetate, isobutyl formate, diethyl ether, di-isopropyl ether, teri-butylmethylether, din-butyl ether, butylvinylether, teri-but
- the liquid or polymeric carrier or matrix material or solution thereof may form the solvent for the salt formation reaction.
- the acid and base components may be added separately to the matrix material or solution thereof, optionally with heating to produce a melt or otherwise ensure a homogenous mixture.
- the product may then be cooled or evaporated and further treated to produce a form suitable for further formulation.
- Suitable ratios of the salt to liquid or polymeric carrier or matrix material may vary from 1 :100 to 10: 1, preferably from 1 :20 to 3:1.
- a volatile solvent used to form the matrix dispersion, it may be difficult to remove it all by evaporation. In the case of solvents such as water or ethanol this is not a problem and substantial residues may be tolerated, indeed may improve the stability and properties of the product. However residues which decrease the viscosity to the extent that crystallisation may occur on storage are undesirable. Less desirable solvents must be removed suffi- ciently by extended, optionally elevated temperature evaporation to ensure a pharmaceutically acceptable product.
- Suitable liquid or polymeric carrier or matrix materials include the following: animal, vegetable or mineral oils, fats, waxes, chocolate, chewing gum base, maize oil, lecithin, groundnut oil, sunflower oil, cottonseed oil, lauroylmonoglyceride, lanolin, gelatin, isinglass, agar, carnauba wax, beeswax, polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), polyethylene glycol esters, ovalbumin, soybean proteins, gum arabic, starch, modified starch, cro- spovidone, hydroxypropyl cellulose, methyl cellulose, carboxymethyl cellulose, sodium car- boxymethyl cellulose, cellulose acetate phthalate, cellulose acetate butyrate, hydroxyethyl cellulose, hydroxypropylmethyl cellulose, ethyl cellulose, chicle, polypropylene, dextrans, in- eluding dexran 40, dextran 70 and dextran
- Preferred materials are PVP and PEG, which are available in various grades differing chiefly in their mean molecular weight. In the case of PVP this may be between 2,000 and 3,000,000, however material in the range 8,000 to 500,000 is more preferred e.g. PVP K-15, K-30, K-60, K-90. In the case of PEG products the mean molecular weight may be in the range 200 to 20,000, but 1,000 to 10,000 is more preferred e.g. PEG 2000, PEG 8000.
- salts of the present invention may be prepared on any suitable scale according to the procedures herein outlined and those procedures which are conventional to one skilled in the art of pharmaceutical chemistry, in particular in the preparation of salt forms. Techniques for scale-up are described in the literature for example Pharmaceutical Process Scale-Up by Michael Levin, Marcel Dekker, New York 2003, ISBN 0824706250, which publication is incorporated herein by reference. Once prepared as described above, amorphous, crystalline and liquid solutions of sildenafil salts with long chain fatty acids may be formulated into pharmaceutical compositions, according to procedures well known in the art.
- Suitable procedures include those provided in Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA, ISBN 0-683-306472; The art, science, and technology of pharmaceutical compounding by Loyd V Allen, American Pharmaceutical Association, 2001, ISBN 1582120358 - incorporated herein by reference.
- these compositions are adapted for oral use such as tablets, capsules, zydis, gums, candies, chocolates, sachet and oral liquids, or are adapted for topical use such as gels, lotions, patches, or ointments, or are adapted for parenteral use such as intravenous, intramuscular, or subcutaneous injection, or are adapted for use as suppositories, or finally are adapted for inhalation therapy such as bronchial or nasal inhalation therapy.
- oral use such as tablets, capsules, zydis, gums, candies, chocolates, sachet and oral liquids
- topical use such as gels, lotions, patches, or ointments
- parenteral use such as intravenous, intramuscular, or subcutaneous injection
- inhalation therapy such as bronchial or nasal inhalation therapy.
- the invention also provides a co-crystal, which co-crystal comprises sildenafil and a co-crystal former, which co-crystal former is a compound which comprises a phenol moiety.
- co-crystal comprises sildenafil
- co-crystal former is a compound which comprises a phenol moiety.
- Compounds which comprise a phenol moiety include phenol itself and phenol substituted with one or more (typically from one to three) ring substituents.
- the co-crystal former is a compound of formula (I)
- n is an integer of from 1 to 3;
- R is H, -C(0)OR 2 , -L-C(0)OR 2 , Ar or -L-Ar;
- R 2 is H or unsubstituted C alkyl
- Ar is phenyl or naphthyl, which phenyl or naphthyl is unsubstituted or substituted with from 1 to 3 groups selected from OH and C alkyl; or
- Ar is a group of formula (II)
- R 3 is H or OH, and m is 0, 1 or 2.
- n 2 or 3.
- R 2 is H or methyl.
- Ar is phenyl, which phenyl is unsubstituted or substituted with from 1 to 3 OH groups; or Ar is typically said group of formula (II) wherein R 3 is OH.
- the co-crystal former may for instance be selected from compounds having the fol
- the co-crystal former is selected from compounds having the following structures:
- the molar ratio of the sildenafil to the co-crystal former in the co-crystal is 1 : 1.
- the co-crystal former is a compound having the following structure:
- the molar ratio of the sildenafil to the co-crystal former in the co-crystal is 3:2.
- the sildenafil in the co-crystal is generally sildenafil base as opposed to a salt of sildenafil, i.e. it is generally 5- ⁇ 2-ethoxy-5-[(4-methylpiperazin-l-yl)sulfonyl]phenyl ⁇ -l- me- thyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one.
- the co-crystal of the invention may for instance be any of the co-crystals exemplified herein below. It may be any one of the co-crystals P42-I-A, P42-I-B, P42-III, P42-VI-B, P42- VI-C, P42-VI-D, P42-VI-E, P42-VII, P42-VIII or P42-IX. It may have the characterisation data of any one of these exemplified co-crystals.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising: (i) a co-crystal of the invention as defined herein, and (ii) a pharmaceutically acceptable carrier.
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichlormethane/methanol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of decanoic acid (0.182 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil decanoate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.68 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 59.4%; H, 7.8%; N, 13.0%
- a solution of sildenafil decanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and decanoic acid (1.82 g) in absolute ethanol (100 ml).
- the solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil decanoate (approx. 0.02 g) is triturated (as described in the main speci- fication) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 59.4%; H, 7.8%; N, 13.0%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication.
- a solution of docosanoic acid (0.36 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil decanoate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.8 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 64.8%; H, 9.2%; N, 10.3%
- a solution of sildenafil docosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and docosanoic acid (3.6 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil docosannoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder dif- fraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta.
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of eicosanoic acid (0.33 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil eicosanoate is isolated as a glass.
- a solution of sildenafil eicosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and eicosanoic acid (3.3 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil eicosanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 64.1%; H, 9.0%; N, 10.7%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of heneicosanoic acid (0.34 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil heneicosanoate is isolated as a glass.
- a solution of sildenafil heneicosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and heneicosanoic acid (3.4 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil heneicosanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta.
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of heptadecanoic acid (0.29 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil heptadecanoate is isolated as a glass.
- the flask After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.79 g. X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material,
- a solution of sildenafil heptadecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and heptadecanoic acid (2.9 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by
- Amorphous sildenafil heptadecanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder dif- fraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 62.9%; H, 8.7%; N, 11.3%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of lauric acid (0.21 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil laurate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.71 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 60.5%; H, 8.1%; N, 12.5% EXAMPLE 17
- a solution of sildenafil laurate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and lauric acid (2.1 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil laurate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 60.5%; H, 8.1%; N, 12.5%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of myristic acid (0.24 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil myristate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.74 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 61.5%; H, 8.3%; N, 12.0%
- a solution of sildenafil myristate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and myristic acid (2.4 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil myristic acid salt (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 61.5%; H, 8.3%; N, 12.0%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of nonadecanoic acid (0.32 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil nonadecanoate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.82 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material. Analysis: C, 63.7%; H, 8.9%; N, 10.9%
- a solution of sildenafil nonandecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and nonadecanoic acid (3.2 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by
- Amorphous sildenafil nonadecanoic acid salt (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 63.7%; H, 8.9%; N, 10.9%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication.
- a solution of nonanoic acid (0.17 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly.
- the solid amorphous sildenafil nonanoate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.67 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 58.8%; H, 7.7%; N, 13.3%
- a solution of sildenafil nonanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and nonanoic acid (1.7 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil nonanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 58.8%; H, 7.7%; N, 13.3%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of octanoic acid (0.152 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly.
- the solid amorphous sildenafil octanoate is isolated as a glass.
- a solution of sildenafil octanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and octanoic acid (1.52 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication.
- a solution of palmitic acid (0.27 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly.
- the solid amorphous sildenafil palmitate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.77 g.
- X-ray pow- der diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 62.4%; H, 8.6%; N, 11.5%
- a solution of sildenafil palmitate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and palmitic acid (2.7 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil palmitate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 62.4%; H, 8.6%; N, 11.5%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of pentadecanoic acid (0.26 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil pentadecanoate is isolated as a glass.
- a solution of sildenafil pentadecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and pentadecanoic acid (2.6 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by
- Amorphous sildenafil pentadecanoic acid salt (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 62.0%; H, 8.4%; N, 11.7%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of stearic acid (0.3 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly.
- the solid amorphous sildenafil stearate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.8 g.
- X-ray pow- der diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 63.3%; H, 8.8%; N, 11.1%
- a solution of sildenafil stearate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and stearic acid (3.0 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil stearate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 63.3%; H, 8.8%; N, 11.1%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication.
- a solution of tetracosanoic acid (0.39 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil tetracosanoate is isolated as a glass.
- a solution of sildenafil tetracosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and tetracosanoic acid (3.9 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication.
- a solution of tricosanoic acid (0.374 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil tricosanoate is isolated as a glass.
- a solution of sildenafil tricosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and tricosanoic acid (3.74 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil tricosanoic acid salt (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 65.2%; H, 9.2%; N, 10.1%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication.
- a solution of tridecanoic acid (0.226 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil tridecanoate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.72 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 61.0%; H, 8.2%; N, 12.2%
- a solution of sildenafil tridecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and tridecanoic acid (2.26 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil tridecanoic acid salt (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 61.0%; H, 8.2%; N, 12.2%
- Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication.
- a solution of undecanoic acid (0.2 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly.
- the solid amorphous sildenafil undecanoate is isolated as a glass.
- the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.7 g.
- X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 60.0%; H, 7.9%; N, 12.7%
- a solution of sildenafil undecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and undecanoic acid (2.0 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
- Amorphous sildenafil undecanoic acid salt (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change.
- Analysis by x-ray powder fraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta.
- EXAMPLE 52 osmotic pump core containing sildenafil citrate
- sildenafil citrate, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- the coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick).
- a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release require- ments.
- EXAMPLE 53 osmotic pump core containing sildenafil free base Active sildenafil 50 mg
- sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid con- sisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid con- sisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- the coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick).
- a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
- EXAMPLE 56 osmotic pump core containing ultrafine sildenafil
- Sildenafil citrate drug substance is loaded into a specially constructed 0.5 metre stainless steel ball milling chamber in which the inner surface has been treated with a Teflon coating.
- the mill cham- ber is then rotated at a speed to allow cascading of mill contents to occur, usually a full rotation every 2 seconds. This milling process is continued for 4 days. After day 4, half of the mannitol is added to chamber and the milling process is continued for a further day.
- the purpose of the mannitol is to act as a carrier substrate for the cohesive ultrafine particles.
- the ultrafine sildenafil, sodium chloride, the remaining mannitol, polyvinylpyrrolidone, and part of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V- blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate for the core are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyeth- ylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- the coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick).
- a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima).
- Typi- cal aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
- the rapid release layer is prepared by blending the ingredients from the rapid release formula table. Sildenafil mesylate, half of the micro crystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in a Fielder granulator for 30 minutes. The mixture is then removed and blended for a further 30 minutes with the remaining dibasic calcium phosphate, sodium croscarmellose, and microcrystalline cellulose using a twin shell V-blender. The magnesium stearate is then added to the mixture and mixed together for 5 minutes.
- This material is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tableting machine to form a rapid release layer around the cores.
- EXAMPLE 58 rapid release layer containing ultrafine sildenafil compressed around an osmotic pump core containing sildenafil citrate
- sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate for the core are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes.
- the dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture.
- the mixture is agitated for 20 minutes or until satisfactory granulation has been achieved.
- the homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator.
- the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyeth- ylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved.
- the coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick).
- a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima).
- Typi- cal aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
- the sildenafil mixture for the rapid release layer is prepared by loading sildenafil citrate drug substance into a specially contracted 0.5 metre stainless steel ball milling chamber in which the inner surface has been treated with a Teflon coating. To the chamber is added a selection of Zirconium Oxide (Zircoa Inc.) grinding beads with diameters ranging from 1 mm up to 50 mm to fill approximately 50% of the chamber volume. The mill chamber is then rotated at a speed to allow cascading of mill contents to occur, usually a full rotation every 2 seconds. This milling process is continued for 4 days. After day 4, half of the microcrystalline cellulose is added to chamber and the milling process is continued for a further day.
- Zirconium Oxide Zircoa Inc.
- microcrystalline cellulose act as a carrier substrate for the cohesive ultrafine particles.
- the milled active is then blended with the remaining microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose in a Fielder a granulator for 30 minutes.
- the mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose in a V-blender for a further 30 minutes.
- the magnesium stearate is then added to the mixture and mixed together for 5 minutes.
- the product is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tablet- ing machine to form the rapid release layer for the drug dosage form.
- EXAMPLE 59 rapid release layer containing an effervescent couple of sildenafil citrate compressed around an osmotic pump core containing sildenafil citrate
- the osmotic pump cores are prepared by the procedures detailed in the preceding Examples 52 onwards.
- the sildenafil mixture for the rapid release layer is prepared by blending the ingredients from the rapid release outer layer formula table.
- the active, half of the microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in a Fielder granulator for 30 minutes.
- the mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose, microcrystalline cellulose in a twin shell V-blender for a further 30 minutes.
- the magnesium stearate is then added to the mixture and mixed together for 5 minutes.
- the product is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tableting machine to form the rapid release layer for the drug dosage form.
- EXAMPLE 60 rapid release layer of sildenafil citrate sprayed around an osmotic pump core containing sildenafil citrate
- the osmotic pump cores are prepared by the procedures detailed in the preceding Examples 52 onwards.
- the rapid release layer of sildenafil is prepared by loading the osmotic pump cores into a Manesty Accela-Cota rotating coating pan and directly film coating the inner cores with a layer of sildenafil citrate mixture until the desired weight gain is achieved.
- Coating is performed using an inlet temperature between 60-66°C, an exhaust temperature between 39- 40°C, a bed temperature fixed between 43-46°C, an atomising air-pressure of 1.5 to 2.5 bar, an airflow of 1400-1500 m 3 /hr (800-900cfm), and a spray rate of 200-300 g/min.
- the sildenafil citrate in the rapid release layer may be replaced by amorphous or ultrafine sildenafil citrate or other salts of sildenafil, for example sildenafil mesylate.
- EXAMPLE 61 capsule containing a rapid release tablet of sildenafil mesylate and an osmotic pump tablet containing sildenafil citrate
- the osmotic core tablet is prepared from the ingredients in the table by the procedures described in the preceding Examples 52 onwards.
- the rapid release tablet is prepared by first blending the ingredients in the rapid release tablet table. Thus, the sildenafil mesylate, half of the microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in a Fielder granulator for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose, microcrystalline cellulose in a twin shell V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and mixed together for 5 minutes. The resulting mixture is then compressed using a Korsch tablet press. The os- motic tablet and the rapid release tablet are encapsulated together into a hard gelation DB capsule Size AA (Capsugel) using a Zanassi 70 capsule filling machine (IMA).
- IMA Zanassi 70 capsule filling machine
- EXAMPLE 62 osmotic pump containing sildenafil citrate with a time delay
- Lubricated granules for proton pump cores are prepared from the ingredients in the table by the procedures described in the preceding Examples 52 onwards.
- the lubricated granules are transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores.
- These cores are then transferred into a Manesty Ac- cela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (80:20) mixture is sprayed on to the cores from a acetone/water mixture until an approximately 20% increase in weight gain has been achieved.
- the coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved.
- a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
- the osmotic pump core is prepared from the ingredients in the table by the procedures de- scribed in the preceding Examples 52 onwards.
- a delay is induced onto the normal operation of the osmotic pump core by the application of a pH dependant coating, for example a coating that will dissolve at pH greater than 5.0.
- a pH dependant coating for example a coating that will dissolve at pH greater than 5.0.
- an enteric coat confers protection to the tablet core until it reaches the small or large intestine.
- a suitable enteric coat may consist of Eudragit L30 D-55 (30% aqueous dispersion) 76.8% w/w, triethyl citrate 7.7 % w/w, talc 15.5 % w/w, or alternatively the enteric coat could consist of Eudragit S100/S12.5.
- EXAMPLE 64 osmotic pump core containing sildenafil citrate with delayed activation, surrounded by a rapid release layer of sildenafil mesylate
- Rapid release outer layer Filler 30 mg microcrystalline cellulose
- Disintegrant 4.25 mg sodium croscarmellose
- the delayed activation osmotic pump core is prepared from the ingredients of the table by the procedures described in Example 62.
- the rapid release layer is prepared by first blending the ingredients in the rapid release outer layer table. All the sildenafil mesylate, half the microcrystalline cellulose, half the dibasic calcium phosphate, and half the sodium croscarmellose are blended in a Fielder granulator for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose, and microcrystalline cellulose using a twin shell V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and mixed together for 5 minutes. This rapid releasing layer is then compressed around the duly formed osmotic pump cores by the employment of a Kilian RUD press-coating tableting machine.
- EXAMPLE 65 osmotic core with an enteric coat containing sildenafil citrate, surrounded by a rapid release layer of sildenafil mesylate
- Rapid release outer layer formula Filler 30 mg microcrystalline cellulose
- Disintegrant 4.25 mg sodium croscarmellose
- the granules are milled and passed through a 20 mesh screen prior to the remainder of the sodium lauryl sulphate being added to the granules and mixed for a further 10 minutes.
- the lubricated granules are then transferred to a conventional tablet press (Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture to approximately 10% increase in weight gain has been achieved.
- the coated cores are then allowed to cure for an appropriate period time and temperature to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick).
- a passage way is created through the semipermeable membrane either through mechanical means or more likely by using a laser drilling machine (Coherent, Convergent Prima).
- Typical aperture sizes can lie between 0.0 5 mm up to 2 mm depending upon therapeutic rate requirements.
- a lag time can be induced on to the osmotic pump by the application of a pH dependant coatings i.e. dissolve at pH's greater than 5.0.
- a pH dependant coatings i.e. dissolve at pH's greater than 5.0.
- the application of a enteric coat guarantees that the coat dissolves either in the small or large intestine where the pH lies be- tween pH 5.0 to 7.0 as oppose to dissolving in the stomach where the pH lies below 5.0.
- the enteric coat consists of Eudragit L30 D-55 (30% aqueous dispersion) 76.8%w/w, Triethyl Citrate 7.7 %w/w, Talc 15.5 % w/w or alternatively the enteric coat could consist of Eudragit S100/S12.5.
- the rapid releasing layer is prepared by first blending the ingredients in the above table. Sildenafil mesylate, half of the microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in fielder granulator for 30 minutes.
- the mixture is then removed and blended with the remaining dibasic calcium phosphate, so- dium croscarmellose, microcrystalline cellulose using a twin shell V-blender for a further 30 minutes.
- the magnesium stearate is then added to the mixture and then mixed for 5 minutes.
- the resulting product is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tableting machine to form the rapid release layer for the drug dosage form.
- sildenafil dosage forms with any combination of rapid, conventional, delayed and prolonged release characteristics to deliver the blood plasma profiles required for the improved formulations of this invention, and to deliver sildenafil within the therapeutic window either more rapidly than the currently marketed swallow tablets containing sildenafil, or continuously for a long period of time compared to the currently marketed swallow tablets containing sildenafil, or more rapidly and continuously for a long period of time when compared to currently marketed swallow tablets containing sildenafil.
- the formulations of the present invention are tested in the animal and human models de- scribed in the patents and references herein mentioned and found to be PDE5 inhibitors.
- the formulations of the present invention can therefore be used in the treatment and/or prophylaxis of the disorders described in the above mentioned patents and are incorporated herein by reference.
- Guidance is to place the tablet under the tongue for 60 seconds (see caffeine summary where we tested how much caffeine dissolved by doing this as substitute for Viagra) then swallow the tablet. Described herein is our model of how the tablet dissolves based on different H levels by our estimates we will deliver the equivalent sildenafil in plasma somewhere around to a normal dose of 100 mg with about 112% to 116% of AUC (so within 505b2 guidelines). We will model and/or test in human next to confirm.
- EXAMPLE 66 Sildenafil Cocrystal Screening
- This Example describes the results of the project entitled “Cocrystal screening of P42” conducted at the Scientific and Technological Centers of the University of Barcelona (CCiT-UB).
- the study has been performed by the technicians of the Polymorphism and Calorimetry Unit (CCiT-UB) in collaboration with the X-ray diffraction Unit (CCiT-UB).
- the main aim of the study was the search of new cocrystals of Sildenafil (referenced as P42) by performing a co- crystal screening.
- Patent EP0463756A1 it claims preparation methods for synthesis of Sildenafil base and other pyrazolopyrimidinethione derivates. PXRD diffractogram and DSC/TGA thermograms are not reported.
- Patent WO02102802A (2002): it claims preparation methods for the synthesis of Sildenafil base and other pyrazolopyrimidinethione derivatives. PXRD diffractogram and DSC/TGA thermograms are not reported.
- Patent WO2007080362A1 it claims Sildenafil Acetylsalicylic acid cocrys- tals. A PXRD diffractogram, crystal data and DSC/TGA thermograms are reported.
- Patent WO2007110559A1 it claims: Sildenafil hydrochloride (polymorphs I, II and III); Sildenafil hydrogensulfate (polymorphs I, II and III); Sildenafil hemi- sulfate (polymorph I); Sildenafil hemitartrate (polymorphs I and II); Sildenafil esyl- ate (polymorphs I, II, III and IV) and Sildenafil fumarate (polymorph I).
- PXRD dif- fractograms, crystal data (sildenafil esylate polymorph I) and a DSC thermogram are reported.
- Patent PT16633364E (2008) preparation methods for the synthesis of Sildenafil base and Sildenafil citrate. PXRD diffractogram and DSC/TGA thermograms are not reported.
- Patent WO2010146407A it claims nanoparticles of Sildenafil citrate. A PXRD diffractogram ( Figure 6) is reported.
- Patent RU2012101818A PXRD diffractogram and DSC/TGA thermograms are not reported.
- P42 is soluble at r.t. in the following solvents: MeOH, EtOH, formic acid, MEK, acetone, MiBK, DMF, DMSO, AcOEt, THF, DME, dioxane, DCM, chloroform, acetic acid, benzyl alcohol and diethylamine; it is soluble at 50°C in IP A, butanol, ACN, toluene, xylene and NH3 (32%) in water. It is insoluble in ethylene glycol, H2O, pentane, heptane, cyclohexane, Et 2 0 and DIE.
- Form P42-E is obtained through slurry experiments by using chloroform as solvent.
- a comparison of the PXRD diagrams of these new forms of P42 is shown in Figures 3 and 4.
- the diagram in Figure 5 summarizes the different behaviour of forms I and II of P42.
- P42-IV P42:Tartaric acid.
- P42-V-A P42:3-Hydroxybenzoic acid. It has been obtained by reaction crystallization in ACN. According to its 'H-NMR analysis, it could be attributed to 1 molecule of 3-hydroxybenzoic acid and 1 ⁇ 4 molecule of ACN per molecule of P42. The PXRD diagram could not be indexed.
- o P42-VI-B It has been obtained by drop grinding in ACN. According to its 'H-NMR analysis, it could be attributed to 1 molecule of 4-hydroxybenzoic acid per molecule of P42. The PXRD diagram could not be indexed, o P42-VI-C: It has been obtained by reaction crystallization in IPA. According to its TGA and 3 ⁇ 4-NMR analysis, it could be attributed to 1 molecule of 4- hydroxybenzoic acid and 1 ⁇ 2 molecule of IPA per molecule of P42. The cell could not be indexed,
- o P42-VI-E It has been obtained by slurry in IPA. According to its TGA and 3 ⁇ 4-NMR analysis, it could be attributed to 1 molecule of 4-hydroxybenzoic acid and 1 ⁇ 4 molecule of IPA per molecule of P42. The cell could not be indexed.
- P42-VII P42:Resorcinol. It has been obtained by slurry in IPA. According to its 3 ⁇ 4- NMR analysis, it could be attributed to 1 molecule of resorcinol per molecule of P42.
- P42-VIII P42:3,4-dihydroxybenzoic acid. It has been obtained by slurry in IPA.
- Salicylic Acid pKa 3.5
- Caffeic Acid pKa 4.6 P42-IX
- P42-A toluene solvate
- P42-B formic acid salt
- P42-C dioxane solvate
- P42-D acetic acid salt or cocrystal
- P42-E hydrate
- the new multicomponent form of P42 with Quercetin (1 :1 stoichiometry) has been obtained in two different forms: one IPA solvate form and one THF solvate form, which have been isolated and characterized.
- the new multicomponent form of P42 with Tartaric acid has been obtained in two different forms: (1 : 1 stoichiometry) and (2: 1 stoichiometry), which have been isolated and characterized. According to the pKa values these forms are expected to be salts.
- the new multicomponent form of P42 with 3 -Hydroxybenzoic acid (1 :1 stoichiometry) has been obtained in ACN solvate form, which have been isolated and characterized. According to the pKa values this form is expected to be a salt.
- the new multicomponent form of P42 with 4-Hydroxybenzoic acid (1 : 1 stoichiometry) has been obtained in two different polymorphic forms, one IPA solvate form and one THF solvate form, which have been isolated and characterized. One of these forms has not been isolated and characterized in pure form. According to the pKa values these forms are expected to be cocrystals.
- the new multicomponent form of P42 with Caffeic acid (2:3 stoichiometry) has been obtained as a hydrated form, which have been isolated and characterized. According to the pKa values this form is expected to be a cocrystal.
- Aerosil 200 (Hydrophilic fumed silica) 1.500 0.500
- the outer layer contains 28.097mg of Sildenafil Citrate per tablet. This is equivalent to 20mg of Sildenafil base.
- Dissolution Summary Aim To produce dissolution profiles at the following conditions for immediate release/sus- tained release development formulations and the comparator product (Pfizer Viagra tablets).
- Dissolution media 0.13 M potassium monophosphate (KH2PO4) adjusted to pH 4.5 with sodium hydroxide
- Dissolution media 50 mM sodium phosphate buffer (NaH 2 P0 4 ), 0.125% CTAB, adjusted to pH 6.8
- sildenafil citrate (equivalent to 20mg
Abstract
The invention provides a pharmaceutical composition comprising sildenafil or a pharmaceutically acceptable salt thereof admixed with excipients in a multicomponent pharmaceutical composition, wherein a first component is adapted to deliver sildenafil rapidly to promote fast onset of action, and a further component is adapted to deliver the sildenafil from dose to dose, wherein the sildenafil is delivered from dose to dose within the therapeutic window.
Description
NEW PHARMACEUTICAL FORMS OF SILDENAFIL
The present invention relates to novel forms and formulations of sildenafil. In particular, the present invention relates to the use of such forms and formulations of sildenafil in the treatment of male sexual dysfunction.
Sildenafil is generically described in US patent 5,250,534 as a selective cGMP PDE inhibitor useful in the treatment of cardiovascular disorders.
Sildenafil free base i.e. 5- {2-ethoxy-5-[(4-methylpiperazin-l -yl)sulfonyl]phenyl}-l- methyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, is specifically described in US patent 6,204,383 Bl as an agent with pharmaceutical utility in the treatment of male sexual dysfunction. Sildenafil free base has the following structure:

We have found phenol co-crystals improve solubility profile of sildenafil to bring about rapid onset of action. The phenol co-crystals improve solubility at all levels of pH therefore improving the performance of the drug during fasted and non- fasted stomach condi- tions.
Example 1 illustrates the preparation of solid sildenafil by the evaporation of a solution in a mixture of dichloromethane and methanol. Examples 2 and 6 describe a solution of sildenafil iodide in dichloromethane, but this is not isolated as a solid product.
The citrate salt of sildenafil (sildenafil citrate) is currently marketed as Viagra® in numerous countries including the USA and the European Union.
The following patents describe various uses, compositions, formulations, and combinations containing sildenafil. These patents are incorporated herein by reference. US Patents
6,743,799, 6,743,443, 6,740,793, 6,740,306, 6,737,070, 6,735,470, 6,734,186, 6,730,786, 6,730,689, 6,730,674, 6,730,505, 6,723,345, 6,713,487, 6,713,295, 6,706,892, 6,706,720, 6,706,691, 6,706,682, 6,706,283, 6,699,991, 6,696,495, 6,696,072, 6,693,122, 6,689,118, 6,686,349, 6,689,338, 6,683,080, 6,682,716, 6,673,987, 6,673,841, 6,673,778, 6,670,386, 6,669,961, 6,667,398, 6,667,060, 6,660,756, 6,656,935, 6,656,452, 6,656,385, 6,650,943, 6,649,606, 6,645,965, 6,645,954, 6,645,528, 6,645,466, 6,644,309, 6,642,274, 6,642,244, 6,638,937, 6,635,638, 6,635,274, 6,634,576, 6,632,419, 6,630,504, 6,627,632, 6,627,234,
6,624,138, 6,623,768, 6,622,721, 6,613,768, 6,613,344, 6,610,747, 6,610,652, 6,605,627, 6,604,698, 6,596,900, 6,596,733, 6,593,369, 6,593,313, 6,592,850, 6,589,990, 6,586,478, 6,585,958, 6,583,147, 6,579,968, 6,579,879, 6,576,653, 6,576,644, 6,573,285, 6,572,880, 6,569,638, 6,569,463, 6,569,143, 6,569,123, 6,566,360, 6,565,851, 6,562,868, 6,562,838, 6,559,184, 6,555,547, 6,552,024, 6,548,544, 6,548,508, 6,548,490, 6,548,087, 6,548,044, 6,544,981, 6,544,563, 6,541,638, 6,541,487, 6,531,297, 6,531,114, 6,528,521, 6,514,536, 6,512,002, 6,511,973, 6,500,610, 6,500,440, 6,499,984, 6,497,885, 6,492,371, 6,492,358, 6,485,747, 6,482,948, 6,482,426, 6,479,493, 6,479,074, 6,477,410, 6,477,410, 6,476,074, 6,476,037, 6,476,021, 6,472,434, 6,472,425, 6,472,420, 6,482,398, 6,469,065, 6,469,024, 6,469,016, 6,469,012, 6,465,494, 6,465,465, 6,462,047, 6,462,044, 6,458,804, 6,458,797, 6,458,790, 6,455,702, 6,455,572, 6,455,654, 6,451,813, 6,451,807, 6,451,339, 6,448,293, 6,444,237, 6,443,152, 6,436,997, 6,436,944, 6,436,684, 6,428,769, 6,426,084, 6,423,683, 6,420,150, 6,417,208, 6,417,207, 6,414,027, 6,413,968, 6,413,496, 6,410,595, 6,410,548, 6,407,259, 6,403,658, 6,403,597, 6,399,601, 6,399,579, 6,395,744, 6,395,736, 6,395,300, 6,391,869, 6,387,407, 6,383,789, 6,383,471, 6,380,267, 6,376,554, 6,376,509, 6,368,640, 6,365,627, 6,365,590, 6,362,178, 6,359,002, 6,350,760, 6,346,271, 6,342,251, 6,338,862, 6,333,354, 6,331,543, 6,326,379, 6,323,242, 6,316,457, 6,316,438, 6,313,164, 6,306,841, 6,303,606, 6,303,135, 6,294,550, 6,294,534, 6,294,192, 6,291,528, 6,291,471, 6,290,986, 6,284,763, 6,277,884, 6,271,228, 6,271,211, 6,268,338, 6,267,985, 6,266,560, 6,265,420, 6,258,373, 6,251,436, 6,251,428, 6,248,363, RE37,234, 6,242,444, 6,241,752, 6,239,117, 6,235,782, 6,235,776, 6,232,321, 6,221,881, 6,221,402, 6,214,849, 6,211,179, 6,211,156, 6,207,829, 6,204,383, 6,200,771, 6,200,591, 6,200,571, 6,197,782, 6,197,778, 6,194,433, 6,187,790, 6,184,231, 6,177,428, 6,172,068, 6,172,060, 6,166,061, 6,165,975, 6,156,753, 6,143,757, 6,143,746, 6,133,272, 6,132,757, 6,130,053, 6,143,757, 6,143,746, 6,133,272, 6,132,757, 6,130,053, 6,127,363, 6,124,461, 6,102,849, 6,100,286, 6,087,368, 6,087,362, 6,077,841, 6,075,028, 6,066,735, 6,051,594, 6,043,252, 6,037,346, 6,013,663, 6,007,824, 5,981,563, 5,958,926, 5,955,611, 5,874,437, 5,858,694 are incorporated herein by reference. In particular all information concerned with preparing the sildenafil active moiety from readily available starting materials is incorporated in full. In addition, it should be appreciated that all information concerning the incorporation of sildenafil into a formulation is herein incorporated by reference as is information on the use of such formulations in medical therapy.
Sildenafil is the active ingredient in Viagra® which is a potent inhibitor of PDE receptors in particular the subtype 5 which is associated with male erectile dysfunction. Sildenafil is a potent muscle relaxant associated with the arterial walls in the cardio vascular system. Sildenafil also antagonises other PDE sub types such as PDE 2 and 6 which are associated with cardiac contractility and blue haze respectively.
Whilst Viagra is a very potent and selective drug its pharmacological profile is far from ideal. It is slow in its absorption through the gut wall and therefore it can take 1 to 2 hours for onset of action since it's delivered in a bolus from a swallow tablet it is absorbed all at once and is then metabolized quickly and expelled from the body leaving a deficit in the blood stream and causing potency to drop and then disappear long before the next dose is due. This is a real problem for suffers of erectile dysfunction trying to plan their sexual encounter to be coincidental with having sufficient medicine in their blood stream but not such a vast excess that unwanted side effects occur such as blue haze and cardiac contractility.
There is a need for a technical solution to this long standing problem and one which solves all three problems at the same time i.e., rapidity, longevity from dose to dose and one which delivers the drug within the therapeutic window between dose to dose. A product line extender having such superior properties would be an obvious commercial success. The fact that no such product has ever been made or described is testament to the fact that there are no obvious solutions to this issue.
A first step in solving this problem is finding variants of the product, i.e. rapidity longevity and maintenance within the therapeutic window from dose to dose which possess physical and chemical characteristics which are conclusive to addressing one or more of the problems associated with PDE 5 inhibition such as rapidity of action duration of effect and sustenance within the therapeutic window for prolonged periods of time from dose to dose.
Co-crystals of many substances are known, co-crystals impart properties of the co- crystal partner (co-crystal former, or co-former) on the target molecule. There is no rational reason why one product forms co crystals whilst others do not even if a target molecule is miss selected it is not possible to pre judge the compounds which would form co crystals and to second guess what those co crystals partners might be. Sildenafil is commercially available as the citrate salt and is therefore a good choice as a starting material. Other salts of sildenafil have been disclosed in the art however none have been tested for rapidity or duration of effect or for any tendency to be within the therapeutic window from dose to dose.
It has been surprisingly found that one class of salts lends itself to forming superior adduct with sildenafil and which adducts possess superior properties. These are the fatty acid salts.
It has also been surprisingly found that specific formulation technologies can be used in conjunction with co-crystals of phenolic compounds and or fatty acids of sildenafil to engineer a superior product possessing superior properties as outlined above, that is to say a more rapidly acting product having a PK profile within the therapeutic window from dose to dose, Information on formulations technologies, co crystals with sildenafil and fatty acid salts of sildenafil are provided herein
Accordingly, the present invention relates to various aspects.
In one aspect, the invention provides co-crystals of sildenafil with a group of phenolic compounds. In particular, the invention provides a co-crystal, which co-crystal comprises sildenafil and a co-crystal former, which co-crystal former is a compound which comprises a phenol moiety. The co-crystal former may be a compound of formula (I) as defined herein.
Another aspect relates to fatty acid salts of sildenafil optionally in the form of co- crystals.
The invention provides a salt of sildenafil with a long chain fatty acid. The salt may be amorphous or crystalline. The long chain fatty acid typically has formula R-C(0)OH wherein R is Ce-24 alkyl or Ce-24 alkenyl.
Another aspect relates to a pharmaceutical composition comprising sildenafil and/or a pharmaceutically acceptable salt or co-crystal thereof admixed with excipients in a multicom- ponent pharmaceutical composition, wherein a first component is adapted to deliver sildenafil rapidly to promote fast onset of action, and a further component is adapted to deliver the sildenafil from dose to dose wherein the sildenafil is delivered from dose to dose within the therapeutic window.
The pharmaceutical composition of the invention may comprise sildenafil and/or a pharmaceutically acceptable salt or co-crystal thereof admixed with excipients in a multicom- ponent pharmaceutical composition, wherein a first component is adapted to deliver sildenafil rapidly to promote fast onset of action, a second component is adapted to deliver the sildenafil from dose to dose, and a third component wherein the sildenafil is delivered from dose to dose within the therapeutic window.
In one embodiment the pharmaceutical composition of the invention comprises a first component which is adapted to deliver the sildenafil rapidly to promote a fast onset of action, and a second component which is adapted to deliver the sildenafil from dose to dose, and which is further adapted to deliver the active ingredient from dose to dose within the therapeutic window. The pharmaceutical composition may be a dosage form, typically a solid dosage form, for instance a swallow tablet.
The invention further provides a pharmaceutical composition comprising sildenafil or a pharmaceutically acceptable salt thereof, which may be as further defined herein (for in- stance, as in the immediately preceding paragraph) wherein the pharmaceutical composition is in the form of a swallow tablet.
Typically the dosage form, for instance a tablet (swallow tablet), comprises two components:
a first component which is adapted to deliver the sildenafil rapidly to promote a fast onset of action, and
a second component which is adapted to deliver the sildenafil from dose to dose, and which is further adapted also to deliver the active ingredient from dose to dose within the therapeutic window.
In the pharmaceutical compositions described herein (including in the dosage forms and swallow tablets) which comprise a first component and one or more further components (for instance a second component, or both a second component and a third component) the first component typically comprises from 10% to 30% by weight, for instance 20% by weight, of the total amount of sildenafil in the composition. The balance of sildenafil is present in the one or more further components. For instance, the balance of sildenafil may be present in a second component, or where second and third components are present, the balance of sildenafil is usually distributed between the second and third components. The total amount of sildenafil in the pharmaceutical compositions described herein (including in the dosage forms and swallow tablets), is often lOOmg, 50mg or 25 mg.
In the pharmaceutical compositions described herein (including in the dosage forms and swallow tablets) which comprise a first component and a second component, the first component typically comprises from 10% to 30% by weight, for instance 20% by weight, of the total amount of sildenafil in the first and second components, and the second component typically comprises from 90% to 70% by weight, for instance 80% by weight, of the total amount of sildenafil in the first and second components. The percentage by weight here refers to the percentage by weight of sildenafil free base which the particular form of sildenafil provides (i.e. weights are quoted on a "free base" basis).
Accordingly, in some embodiments, the total amount of sildenafil in the first and second components (which again refers to the total amount of sildenafil free base which the particular form or forms of sildenafil in the first and second components provide) is 100 mg, and the first component comprises from lOmg to 30mg of sildenafil and the second component comprises from 90mg to 70mg of sildenafil. The first component may for instance comprise 20mg of sildenafil and the second component may comprise 80mg of sildenafil.
In other embodiments, the total amount of sildenafil in the first and second components is 50 mg, and the first component comprises from 5mg to 15mg of sildenafil and the second component comprises from 45mg to 35mg of sildenafil. The first component may for instance comprise 1 Omg of sildenafil and the second component may comprise 40mg of sildenafil.
In other embodiments, the total amount of sildenafil in the first and second components is 25 mg, and the first component comprises from 2.5mg to 7.5mg of sildenafil and the second component comprises from 17.5mg to 22.5mg of sildenafil. The first component may for instance comprise 5mg of sildenafil and the second component may comprise 20mg of sildenafil.
The sildenafil in the first component may be sildenafil free base. Alternatively, the sildenafil in the first component may be a pharmaceutically acceptable salt of sildenafil. It may for instance be sildenafil citrate. Alternatively, it may be a fatty acid salt of sildenafil. In another embodiment, the sildenafil in the first component may be in the form of a co-crystal of sildenafil. For all these forms of sildenafil, the dosage of sildenafil, quoted on a free base basis, may be as defined above.
The sildenafil in the second component is often a pharmaceutically acceptable salt of sildenafil. Sildenafil citrate is one preferred choice for the second component. Alternatively, the sildenafil in the second component may be a fatty acid salt of sildenafil. In another em- bodiment, the sildenafil in the second component is sildenafil free base. In yet another embodiment, the sildenafil in the second component is in the form of a co-crystal of sildenafil, for instance a co-crystal of sildenafil with a compound which comprises a phenol moiety. For all these forms of sildenafil, the dosage of sildenafil, quoted on a free base basis, may be as defined above.
The first component typically comprises from lOmg to 35mg, for instance from lOmg to 30mg, or for instance from 20mg to 25mg, of sildenafil. The first component may for instance comprise 20mg of sildenafil. The first component may alternatively comprise 25 mg of sildenafil. The sildenafil in the first component may be sildenafil free base. Alternatively, the sildenafil in the first component may be a pharmaceutically acceptable salt of sildenafil. It may for instance be sildenafil citrate. Alternatively, it may be a fatty acid salt of sildenafil. In another embodiment, the sildenafil in the first component may be in the form of a co-crystal of sildenafil. For all these forms of sildenafil, the dosage of sildenafil, quoted on a free base basis, may be as defined above.
The second component typically comprises from 40mg to 1 OOmg of sildenafil, for in- stance from 40mg to 90mg of sildenafil, or, for example, from 50 mg to 80 mg of sildenafil. The second component may for instance comprise 80mg of sildenafil. The second component may alternatively comprise 50mg of sildenafil. The second component in some embodiments comprises from 70mg to 90mg of sildenafil, for example 80mg of sildenafil. The sildenafil in the second component is often a pharmaceutically acceptable salt of sildenafil. Sildenafil cit- rate is one preferred choice for the second component. Alternatively, the sildenafil in the second component may be a fatty acid salt of sildenafil. In another embodiment, the sildenafil in the second component is sildenafil free base. In yet another embodiment, the sildenafil in the second component is in the form of a co-crystal of sildenafil, for instance a co-crystal of sildenafil with a compound which comprises a phenol moiety. For all these forms of sildena- fil, the dosage of sildenafil, quoted on a free base basis, may be as defined above.
Thus, the first component may comprise from lOmg to 35mg of sildenafil and the second component may comprise from 40mg to lOOmg of sildenafil. The first component may
for instance comprise from lOmg to 30mg of sildenafil, and the second component may comprise from 40mg to 90mg of sildenafil. The first component may for instance comprise from 20mg to 25mg of sildenafil, and the second component may comprise from 50mg to 80mg of sildenafil. The first component may comprise about 20mg of sildenafil and the second compo- nent may comprise about 80mg sildenafil. Alternatively, the first component may comprise about 25mg of sildenafil and the second component may comprise about 50mg sildenafil.
The first component may be adapted to deliver sildenafil rapidly, wherein the adaptation is that the first component comprises sildenafil admixed with excipients in a manner which promotes rapid, preferably immediate, release of the sildenafil.
The first component may comprise sildenafil admixed with excipients, which may be conventional excipients, which promote immediate release.
The first component may for instance comprise from lOmg to 35mg, for example from lOmg to 30mg, or from 20mg to 25 mg, of sildenafil with such excipients which promote immediate release. It may for instance comprise 20mg, or 25mg, of sildenafil with such excipients which promote immediate release. The excipients may be conventional excipients. The sildenafil in the first component may be sildenafil free base. Alternatively, the sildenafil in the first component may be a pharmaceutically acceptable salt of sildenafil. It may for instance be sildenafil citrate. Alternatively, it may be a fatty acid salt of sildenafil. In another embodiment, the sildenafil in the first component may be in the form of a co-crystal of sildenafil. For all these forms of sildenafil, the dosage of sildenafil, quoted on a free base basis, may be as defined above.
The second component of the pharmaceutical composition of the invention may comprise sildenafil with excipients, for instance conventional excipients, which promote modified release.
The second component may for instance comprise from 40mg to lOOmg, for example from 40mg to 90mg, or from 50mg to 80mg, of sildenafil with such excipients which promote modified release. It may for instance comprise 50mg, or 80mg, of sildenafil with such excipients. The second component may for instance comprise from 70mg to 90mg, of sildenafil, for instance 80mg sildenafil citrate, with such excipients. The excipients may be conventional ex- cipients. The excipients which promote modified release in the second component may be polymers. The sildenafil in the second component is often a pharmaceutically acceptable salt of sildenafil. Sildenafil citrate is one preferred choice for the second component. Alternatively, the sildenafil in the second component may be a fatty acid salt of sildenafil. In another embodiment, the sildenafil in the second component is sildenafil free base. In yet another em- bodiment, the sildenafil in the second component is in the form of a co-crystal of sildenafil, for instance a co-crystal of sildenafil with a compound which comprises a phenol moiety. For
all these forms of sildenafil, the dosage of sildenafil, quoted on a free base basis, may be as defined above.
Another aspect of the invention provides sildenafil adapted in a multi component dosage form, such as a tablet, said dosage form being adapted in a first way to provide rapid re- lease of sildenafil into the bloodstream, said dosage form being adapted in a second way to further provide a maintenance dose of sildenafil within the therapeutic window, and the dosage form being adapted in a third way to provide a modified or delayed release format of the sildenafil product which lasts from dose to dose.
The exact dose of each component is dependent on the choice of formulation but in general the choice of each component will be made so that the amount of active ingredient in each component delivers the right amount of drug product to ensure rapidity, longevity from dose to dose and maintenance of dose within the therapeutic window.
As a rule of thumb the total amount of sildenafil in a formulation is at least 20mg and usually 50mg to lOOmg.
A preferred aspect of the invention is a product (usually a multi component dosage form, such as a tablet) which comprises:
sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co- crystal is optionally as further defined herein;
a fatty acid salt of sildenafil, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and a delayed release component, comprising a bolus dose of sildenafil. Often, the sildenafil in the bolus dose is in the form of sildenafil citrate.
Another preferred aspect of the invention is a product (usually a multi component dosage form, such as a tablet) which comprises:
from 2.5mg to 25mg sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co-crystal is optionally as further defined herein;
from 2.5mg to 25mg sildenafil in the form of a fatty acid salt of sildenafil as defined herein, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and
said delayed release component, comprising a bolus dose of sildenafil. The bolus dose of sildenafil may be from 20mg to 95mg sildenafil. The sildenafil in the bolus dose may be in the form of sildenafil citrate.
Another preferred aspect of the invention is a product (usually a multi component dosage form, such as a tablet) which comprises:
from 2.5 to 5mg sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co-crystal is optionally as further defined herein;
from 2.5 to 15mg sildenafil in the form of a fatty acid salt of sildenafil as defined herein, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and
said delayed release component, comprising a bolus dose of sildenafil. The sildenafil in the bolus dose may be in the form of sildenafil citrate. The bolus dose may for example be from lOmg to 80mg sildenafil, for instance 20mg sildenafil.
Another preferred aspect of the invention is a product (usually a multi component dosage form, such as a tablet) which contains: 5mg of sildenafil co- crystals with a phenol to deliver rapidity, 15mg of sildenafil fatty acid adapted using time release technology to deliver sildenafil between onset of action and kick in of a bolus amount of delayed release component, and a bolus amount of delayed release component of sildenafil which is suitably 20mg.
Except where specified otherwise, all sildenafil doses are cited on a free base basis. Thus if the sildenafil is in the free base form, the term "80mg sildenafil", as used herein, refers to 80mg sildenafil free base. However, if the sildenafil is in the form of the citrate salt, the term "80mg sildenafil" as used herein means 112mg of sildenafil citrate (if the mass of the sildenafil citrate is rounded to zero decimal places). Similarly, if the sildenafil is in the free base form, the term "20mg sildenafil", as used herein, refers to 20mg sildenafil free base. However, if the sildenafil is in the form of the citrate salt, the term "20mg sildenafil" as used herein means 28 mg of sildenafil citrate (if the mass of sildenafil citrate is rounded to zero decimal places).
Brief Description of the Figures
Fig. 1 shows powder X-ray diffractograms of the initial batch of sildenafil (P42) as received (upper diffractogram) and the published P42 form: QEGTUT anhydrous form (lower diffractogram).
Fig. 2 shows powder X-ray diffractograms of the initial P42 batch as received (upper diffractogram) and after grinding (lower diffractogram).
Fig. 3 shows powder X-ray diffractograms of P42 polymorphs P42 Form I (upper diffractogram) and P42 Form II (lower diffractogram).
Fig. 4 shows powder X-ray diffractograms of P42 polymorphs P42-A (first and top diffractogram), P42-B (second diffractogram), P42-C (third diffractogram), P42-D (fourth diffractogram) and P42-E (fifth and bottom diffractogram).
Fig. 5 is a diagram summarising the different behaviours of forms of P42.
Fig. 6 shows powder X-ray diffractograms of the following new multicomponent forms of P42: P42-I-A (first and top diffractogram), P42-I-B (second diffractogram), P42-III (third diffractogram), P42-IV-A (fourth diffractogram), P42-IV-B (fifth diffractogram), P42- V-A (sixth and bottom diffractogram).
Fig. 7 shows powder X-ray diffractograms of the following new multicomponent forms of P42: P42-VI-B (first and top diffractogram), P42-VI-C (second diffractogram), P42- VI-D (third diffractogram), P42-VI-E (fourth diffractogram), P42-VII (fifth diffractogram), P42-VIII (sixth diffractogram) and P42-IX (sixth and bottom diffractogram).
Fig. 8 shows the dissolution profile of 25mg Viagra tablets in 0.01M hydrochloric acid.
Fig. 9 shows the dissolution profile of 50mg Viagra tablets in 0.01M hydrochloric acid.
Fig. 10 shows the dissolution profile of lOOmg Viagra tablets in 0.01M hydrochloric acid.
Fig. 11 shows the dissolution profile in 0.01M hydrochloric acid of the sustained-release core development formulation no. 16CF25/026 comprising sildenafil citrate, as described in Example 67.
Fig. 12 shows the dissolution profile in 0.01M hydrochloric acid of the immediate - release development formulation no. 16CF25/027 comprising sildenafil citrate, as described in Example 67.
Fig. 13 shows the dissolution profile of 25mg Viagra tablets in pH 4.5 phosphate buffer.
Fig. 14 shows the dissolution profile of 50mg Viagra tablets in pH 4.5 phosphate buffer.
Fig. 15 shows the dissolution profile of lOOmg Viagra tablets in pH 4.5 phosphate buffer.
Fig. 16 shows the dissolution profile in pH 4.5 phosphate buffer of the sustained-release core development formulation no. 16CF25/026 comprising sildenafil citrate, as de- scribed in Example 67.
Fig. 17 shows the dissolution profile in pH 4.5 phosphate buffer of the immediate -release development formulation no. 16CF25/027 comprising sildenafil citrate, as described in Example 67.
Fig. 18 shows the dissolution profile of 25mg Viagra tablets in pH 6.8 phosphate buffer with 0.125% CTAB.
Fig. 19 shows the dissolution profile of 50mg Viagra tablets in pH 6.8 phosphate buffer with 0.125% CTAB.
Fig. 20 shows the dissolution profile of lOOmg Viagra tablets in in pH 6.8 phosphate buffer with 0.125% CTAB.
Fig. 21 shows the dissolution profile in pH 6.8 phosphate buffer with 0.125% CTAB of the sustained-release core development formulation no. 16CF25/026 comprising sildenafil citrate, as described in Example 67.
Fig. 22 shows the dissolution profile in pH 6.8 phosphate buffer with 0.125% CTAB of the immediate -re lease development formulation no. 16CF25/027 comprising sildenafil citrate, as described in Example 67.
Fig. 23 shows the dissolution profile in 0.01M hydrochloric acid of the immediate - release development formulation no. 16CF25/033 comprising sildenafil free base, as described in Example 68.
Fig. 24 shows the dissolution profile in pH 4.5 phosphate buffer of the immediate -release development formulation no. 16CF25/033 comprising sildenafil free base, as described in Example 68.
Detailed Description of the Invention
Information on how to make and use the invention is provided herein. The references mentioned herein are also incorporated herewith. The pharmacological test methods are also incorporated.
The term "alkyl", as used herein, refers to a linear or branched chain saturated hydrocarbon radical. A "Cn-m alkyl" refers to an alkyl having from n to m carbon atoms. Thus, an alkyl group may be a Ci-25 alkyl group, a Ci-24 alkyl group, a Ce-24 alkyl group, a Ci-10 alkyl group, a Ci-6 alkyl group or a C1-4 alkyl group. Examples of a Ci-10 alkyl group are methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl or decyl. Examples of Ci-6 alkyl groups are methyl, ethyl, propyl, butyl, pentyl or hexyl. Examples of C1-4 alkyl groups are methyl, ethyl, i-propyl, n-propyl, t-butyl, s-butyl or n-butyl. If the term "alkyl" is used without a prefix specifying the number of carbons anywhere herein, it has from 1 to 4 carbons.
The term "alkenyl", as used herein, refers to a linear or branched chain hydrocarbon radical comprising one or more double bonds. A "Cn-m alkenyl" refers to an alkenyl having from n to m carbon atoms. Thus, an alkenyl group may be a C2-25 alkenyl group, a C2-24 alkenyl group, a Ce-24 alkenyl group, a C2-10 alkenyl group, a C2-6 alkenyl group or a C2-4 alkenyl group. Examples of a C2-10 alkenyl group are ethenyl (vinyl), propenyl, butenyl, pen- tenyl, hexenyl, heptenyl, octenyl, nonenyl or decenyl. Examples of C2-6 alkenyl groups are ethenyl, propenyl, butenyl, pentenyl or hexenyl. Examples of C2-4 alkenyl groups are ethenyl, i-propenyl, n-propenyl, s-butenyl or n-butenyl. Alkenyl groups typically comprise one or two double bonds.
The term "co-crystal" (or "co crystal" or "cocrystal") as used herein means a solid that is a crystalline single phase material comprising two or more different molecular and/or ionic compounds which are neither solvents nor simple salts (S. Aitipamula et al. "Poly- morphs, Salts, and Cocrystals: What's in a Name?", Cryst. Growth Des., 2012, 12 (5), pp
2147-2152). The two or more different molecular and/or ionic compounds in a co-crystal are generally compounds which are themselves solid at room temperature (i.e. solids at 22 °C).
They are typically present in the co-crystal in a definite stoichiometric ratio. In the co-crystals of the present invention, one of the two or more different molecular and/or ionic compounds is an API, sildenafil, and another of the two or more different molecular and/or ionic compounds is a co-crystal former. Indeed, the co-crystals of the present invention are pharmaceu- tical co-crystals. A pharmaceutical co-crystal is a crystalline single phase material comprising an API and one or more unique co-crystal formers, typically in a stoichiometric ratio. Each of the one or more co-crystal formers is a molecular or an ionic compound that is a solid at room temperature. Solvates (including hydrates) of an API that do not further comprise a co-crystal former are therefore not considered to be co-crystals. A pharmaceutical co-crystal may how- ever include one or more solvent (e.g. acetonitrile, or water) molecules in the crystal lattice which comprises the API and the one or more unique co-crystal formers. Co-crystals can be constructed through several types of interaction, including hydrogen bonding (H -bonding), pi stacking, and van der Waals forces. However, co-crystals often rely on hydrogen-bonded assemblies between neutral molecules of an API and another (co-crystal former) component.
The dosage forms of the present invention are proven to work as pde5 inhibitors and find use in the treatment of male erectile dysfunction. Fresh frozen human penis was obtained from HAM (Pennsylvania). Tissue was thawed at room temperature, the corpus cavernosum was dissected from the penis to yield approximately 2-4 g of tissue and the following isolation protocol was followed. Tissue was coarsely chopped in ice-cold isotonic buffer (35 ml) con- taining 250 mm sucrose, 1 mM EDTA, 0.5 mM PMSF and 20 mM HEPES, pH 7.2, and the mixture subjected to brief (1 min.) treatment with a Silversen mixer/emulsifier. Homogenates were prepared using homogeniser tubes with Teflon pestles and a soluble fraction was prepared by centrifugation at 100,000 x g for 60 min. at 4°C. 10 ml of high speed supernatant was applied to a Pharmacia Mono Q anion exchange column (1 ml bed volume) equilibrated with buffer containing 1 mM EDTA, 0.5 mM PMSF and 20 mM HEPES, pH 7.2 (chromatography buffer). The column was then washed with 5 bed volumes of chromatography buffer, after which PDEs were eluted using a continuous gradient of 0-500 mM NaCl (total volume 35 ml) and 1 ml fractions collected.
Column fractions were assayed for PDE activity using 500 nM cGMP or 500 nM cAMP as substrate. cAMP PDE activity was also determined in the presence of 1 μΜ unlabeled cGMP and the PDE activity of selected fractions was determined in the presence of 10 mM CaCh and 10 units/ml bovine brain calmodulin. Appropriate fractions were pooled and stored at 4°C during the course of the study.
Inhibition studies were performed using a substrate concentration of 500 nM through- out. All inhibitors were dissolved in DMSO and concentration-response curves were constructed over the range 3xl0"10 to lxl0"4 M in half log increments. IC50 values were calculated using the sigmoidal curve fitting algorithm of bio stat.
Human corpus cavernosum soluble PDEs were separated into three distinct fractions of activity. The first, fraction I, (designated by order of elution) represented the major PDE present and was highly selective for cGMP as substrate. This fraction was found to be insensitive to stimulation by calcium/calmodulin and was classified as PDEv. Fraction II hydro lysed cGMP and cAMP, with the latter activity being stimulated in the presence of cGMP, and was classified as PDEn, whilst fraction III was cAMP selective and this activity was inhibited in the presence of cGMP, consistent with PDEm activity.
In order to further characterise the PDE isoenzymes present in the tissue, studies were performed using a variety of inhibitors. Inhibitor studies with fractions I and II were per- formed using cGMP as substrate, whilst fraction III studies utilised cAMP. These studies confirmed that fraction I corresponds to PDEv, whilst fraction III was clearly Identified as PDEm ; fraction II (PDEn) was relatively insensitive to all the inhibitors tested.
In summary, the above investigation identified three PDE isoenzymes in human corpus cavernosum tissue. The predominant PDE was the cGMP-specific PDEv, whilst cGMP- stimulated cAMP PDEn and cGMP-inhibited cAMP PDEm were also present.
The formulations of the present inventions are tested in vitro and found to be potent and selective inhibitors of the cGMP-specific PDEv. Thus relaxation of the corpus cavernosum tissue and consequent penile erection is presumably mediated by elevation of cGMP levels in the said tissue, by virtue of the PDE inhibitory profile of the compounds of the inven- tion.
The sildenafil summary shows the in vitro dissolution profiles for the two elements of the formulation, the inner sustained release core with 80mg sildenafil and the outer core which is an ODT with 20mg sildenafil. Guidance is to place the tablet under the tongue for 60 seconds (see caffeine summary where we tested how much caffeine dissolved by doing this as substitute for Viagra) then swallow the tablet. Described herein is our model of how the tablet dissolves based on different pH levels We assume 20% will go into blood plasma PK from the sublingual element (some will go normal route so even if 30% dissolves not all will go sublingual). This would give us 4mg sublingual which according to other papers could deliver 4 to 5 times the dose in systemic circulation than normal, i.e. same amount as 16-20mg of normal citrate but importantly much quicker, i.e. 15mins. The rest of the tablet will be swallowed and therefore all the ODT dissolves in the stomach so the blood plasma PK will be as Pfizer ODT data the core will then dissolve slowly in the intestine. By our estimates we will deliver the equivalent sildenafil in plasma somewhere around to a normal dose of 100 mg with about 112% to 116% of AUC (so within 505b2 guidelines) modelling and or testing in humans will confirm. A two component tablet is preferred the sildenafil summary shows the in vitro dissolution profiles for the two elements of the formulation.
The present invention relates to novel pharmaceutical formulations, in particular to modified release formulations for the delivery of a therapeutically useful amount of sildenafil. Sildenafil citrate (Viagra®) is currently approved for the treatment of erectile dysfunction in male humans. Sildenafil is generically described in US patent 5,250,534 as a selective cGMP PDE inhibitor useful in the treatment of cardiovascular disorders such as angina, hypertension, heart failure and atherosclerosis. Sildenafil is specifically described in US patent 6,204,383 Bl as an agent with pharmaceutical utility in the treatment of male sexual dysfunction (MED).
The physiological mechanism of erection of the penis involves release of nitric oxide (NO) in the corpus cavernosum during sexual stimulation. (NO) then activates the enzyme guanylate cyclase, which results in increased levels of cyclic guanosine monophosphate (cGMP), producing smooth muscle relaxation in the corpus cavernosum and allowing inflow of blood. Sildenafil has no direct relaxant effect on isolated human corpus cavernosum, but enhances the effect of nitric oxide by inhibiting phosphodiesterase type5 (PDE5), which is re- sponsible for degradation of cGMP in the corpus cavernosum. When sexual stimulation causes local release of (NO), inhibition of PDE5 by sildenafil causes increased levels of cGMP in the corpus cavernosum, resulting in smooth muscle relaxation and inflow of blood to the corpus cavernosum. Sildenafil at recommended doses has no effect in the absence of sexual stimulation.
Sildenafil citrate in currently marketed swallow tablets is available in three dosage forms i.e. 25 mg, 50 mg and 100 mg of sildenafil calculated as the free base. For currently marketed swallow tablets, the time for maximum plasma concentrations of sildenafil is between 0.5 and 2 hours when the patient is in a fasting state, but is delayed, on average, by 1 hour when sildenafil is taken with a high fat meal. After the initial variable delay in the onset of action, a therapeutic effect is maintained for approximately 2 hours, followed by a diminished response for about a further 2 hours.
When administering a conventional swallow tablet from which the active moiety is absorbed into the bloodstream, there is usually a minimum blood plasma concentration which is required to achieve a therapeutic effect. As the blood plasma level increases, so does the therapeutic effect in a dose-related manner until a maximal therapeutic effect has been achieved. However, when drug blood plasma levels exceed a certain concentration, undesired side-effects become apparent. When drug blood plasma levels drop below a certain concentration there is little or no therapeutic benefit. Drug blood plasma concentrations between these two levels are often referred to as the therapeutic window. Hence, blood plasma levels outside the therapeutic window are associated with either a lack of efficacy or unacceptable side-effects. For example, it has been reported that high doses of sildenafil can result in the inhibition of other phosphodiesterases, for example PDE 2 and PDE 6. PDE2 has been linked
to the control of cardiac contractility, whereas PDE6 is found in the retina and is involved in the phototransduction pathway of the retina. A common side-effect of sildenafil when delivered by currently marketed swallow tablets is "blue haze", i.e. a transient dose-related impairment of colour discrimination, which is consistent with PDE6 inhibition.
Warnings are provided on package inserts for existing sildenafil products against administering sildenafil to patients with pre-existing cardiac conditions.
In summary, currently approved swallow tablets have a number of draw-backs. For example, they suffer from a slow and variable onset of action (i.e. they do not achieve the lower limit of the therapeutic window for a significant and variable period of time). Also, currently approved swallow tablets have a tendency to produce excessive concentrations of sildenafil (i.e. blood plasma levels may exceed the upper limit of the therapeutic window).
In conclusion, currently approved swallow tablets suffer from a slow and variable onset of action, a short duration of action, and a risk of unacceptable side-effects.
Sildenafil free base i.e. 5-[2-ethoxy-5-(4-methylpiperazin-l -ylsulfonyl)phenyl]-l- methyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, is specifically described in US patent 6,204,383 Bl . Example 1 illustrates the preparation of solid sildenafil by the evaporation of a solution in a mixture of dichloromethane and methanol. Examples 2 and 6 describe a solution of sildenafil iodide in dichloromethane, but this is not isolated as a solid product.
Sildenafil is currently marketed as the citrate salt in numerous countries including the
USA and the European Union.
Sildenafil citrate is the active ingredient in VIAGRA®, which is approved for use in the treatment of MED. The choice of an improved formulation of sildenafil to deliver a clinically optimal dose of sildenafil is not a matter of routine. Other PDE5 agents have recently become available on the market for the treatment of MED. These agents include: Cialis®, and Levitra®. These agents have a faster onset of action and longer duration when compared to Viagra®, thereby emphasising the current deficiencies of Viagra®.
In view of the above, there is a clear need to find alternative formulations of sildenafil as compared to the currently marketed form, which deliver a more rapid onset of therapeutic efficacy and a prolonged duration of therapeutic efficacy within the therapeutic window. Furthermore, there is a need to provide alternative formulations that do not depend so markedly on fasting for a more rapid onset of therapeutic efficacy when compared to the currently marketed product, and which are more suited for use in a relaxed environment, which is appropriate to non-clinical sexual activity.
Unlike many other medical conditions, sexual dysfunction is greatly affected and indeed potentiated by stress and other factors including anxiety, uncertainty, time-
restrictions, the need for a controlled or limited diet and a clinical environment.
Hence, maximum benefit for the patient is achieved when therapy takes place in as natural an environment as possible, when there is no pressure to keep to a strict timetable as a result of a narrow time-window for therapeutic efficacy, and when there is freedom from distracting side-effects.
The time interval between dosing and onset of therapeutic efficacy with currently marketed swallow tablets is between 0.5 and 2 hours for fasted healthy male volunteers, and is delayed by a median 60 minutes following a high fat meal. In the general population, this uncertainty is likely to be even greater, and represents a major disadvantage for a stress-free sexual environment. Hence there is a need for a formulation which provides a rapid and predictable onset of therapeutic effect.
The duration of therapeutic efficacy for conventional swallow tablets is very short and not conducive to relaxed sexual activity. In real-life situations, the opportunity for sexual activity may not always be predictable with any degree of certainty, so the need for strict time-tables represents a major disadvantage which may cause high levels of counter-productive stress and anxiety. Hence there is a need for a formulation that delivers a therapeutically effective blood plasma level of sildenafil for longer than 2 hours and for up to about 24 hours.
The sharp bolus of sildenafil released into the blood plasma by currently marketed tablets, as described in the art, is undesirable because of the risk of side-effects. The target population for therapy includes many people of above middle age who are potentially vulnerable to side-effects associated with cardiac contractility, and who may be tempted to increase the dose beyond the recommended level. Furthermore, even clinically non-significant side- effects such as temporary vision impairment represent unwanted distractions which may increase stress and anxiety.
We have surprisingly found that sildenafil formulations can be prepared which provide either a rapid and predictable onset of therapeutic efficacy, or a rapid and predictable onset of therapeutic efficacy combined with a prolonged duration of therapeutic efficacy and a control of excessive blood plasma levels and adverse events. Such formulations are less affected by fasting and are more suited for use in a relaxed environment which is appropriate to non-clinical sexual activity when compared with the conventional swallow tablets of the prior art.
Whenever mentioned herein, the term predictability is to be understood to mean that the patient should have confidence in the achievement of sufficient sildenafil blood plasma concentrations necessary for the therapeutic effect that enables sexual activity when the op- portunity for sexual activity arises, which opportunity inevitably cannot be timed exactly.
Accordingly, the present invention provides a modified release drug delivery dosage form which delivers sildenafil within the therapeutic window either more rapidly than the currently marketed swallow tablets containing sildenafil, or continuously for a long period of time compared to the currently marketed swallow tablets containing sildenafil, or more rap- idly and continuously for a long period of time when compared to currently marketed swallow tablets containing sildenafil. Suitably, the modified release formulation of the present invention is a rapid release, or a delayed release, or a prolonged release formulation, or any combination of the aforementioned, optionally combined with a conventional release component.
One published pharmacokinetic study, which is described in the prescribing infor- mation associated with the currently marketed swallow tablets, quotes mean sildenafil blood plasma levels in healthy human male volunteers. Hence, as a starting point, one may calculate the blood plasma levels of sildenafil which fall within the therapeutic window in an average human male patient suffering from MED, but otherwise healthy, to be about 50 ng/ml to 500 ng/ml, probably 100 ng/ml to 400 ng/ml, more probably 150 ng/ml to 350 ng/ml, and even more probably 175 ng/ml to 350 ng/ml and most probably between 200 to 325 ng/ml.
But, it should be appreciated that interpatient variability results in the therapeutic window varying from patient to patient, and hence in the need for individual dose adjustment in order to achieve the optimal benefits that the present invention provides.
It is clear from all the above information, that the therapeutic window for sildenafil may also be defined in terms of efficacy and side-effect profile. The lower threshold of the therapeutic window is equivalent to the threshold at which an adequate improvement in erectile function is delivered. The higher threshold of the therapeutic window is the threshold at which side-effects become unacceptable. It is apparent that the therapeutic window lies between the thresholds at which a substantially undiminished erectile response is achieved.
It is therefore a feature of this invention that the formulations of this invention are provided in a series of dosing strengths for the purposes of dose titration, i.e. they permit an individual patient to commence with a low dose formulation and increase the dose systematically until an optimal effect is achieved. It is a further feature of this invention that the formulations of this invention are designed to provide a release of sildenafil within the above-de- fined therapeutic window.
Whenever mentioned herein the term sildenafil is to be understood to include sildenafil in the form of the free base and also in the form of a pharmaceutically acceptable salt. Sildenafil free base is the compound 5-[2-ethoxy-5-(4-methylpiperazin-l-ylsulfonyl) phenyll- methyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one which is the active moiety in Viagra™. The term "pharmaceutically acceptable salt thereof refers to salts which are physically, chemically and physiologically acceptable for either human or veterinary use.
It should also be understood that sildenafil or a pharmaceutically acceptable salt thereof includes solutions, amorphous forms, and crystalline forms of sildenafil including solvates, hydrates, co-crystals and polymorphs.
Whenever a scientific or patent reference is quoted herein it must be understood that such reference is incorporated herein by reference in full including other references quoted therein.
The term "more rapidly", in the context of rapid release formulations, in the context of this specification, means that a therapeutic blood plasma concentration of sildenafil is achieved more rapidly than with the swallow tablets of the prior art, particularly in the non- fasted state, and more particularly when taken with a high fat meal.
The term "continuously for a long period of time" in the context of this specification means that sildenafil should be released within the therapeutic window over a period of more than 2 and up to 24 hours, for example between 2 hours and 5 hours, preferably between 2 and 10 hours, and more preferably between 0.15 and 24 hours (i.e. from dose to dose).
It should be understood that when the terms "more rapidly" and "continuously for a long period of time" are used together as "more rapidly and continuously for a long period of time" the release of sildenafil should bridge the complete period of time between the onset of therapeutic efficacy implied by the term "more rapidly" and the diminution of the sildenafil blood plasma concentration below the therapeutic window implied by the term "continuously for a long period of time".
The term "rapid release", in the context of "modified release", in the context of this specification, means a formulation by means of which a therapeutic blood plasma concentration of sildenafil is achieved more rapidly than with the swallow tablets of the prior art, particularly in the non-fasted state, and more particularly when taken with a high fat meal.
The term 'delayed release', in the context of "modified release", in the context of this specification, is understood to indicate a formulation that is designed to retard the initial release of drug from the dosage form by a pre-determined interval of time. In the case of sildenafil, delayed release may be understood to mean retardation of release, when compared to the currently approved product which is described as releasing sildenafil between 0.15 and 2 hours when measured in the fasting state, and one hour longer when taken with a high fat meal.
The term "prolonged release", in the context of "modified release", in the context of this specification, may be understood to indicate a formulation that is designed to maintain the release of drug over a period of time that is substantially greater than is achieved in the cur- rently marketed formulation. In the case of sildenafil prolonged release, substantially greater means that the drug is released within the therapeutic window for longer than 2 hours with no
marked diminution of therapeutic effect. Accordingly, sildenafil is released within the therapeutic window over a period of more than 2 and up to 24 hours, for example between 2 hours and 5 hours, preferably between 2 and 10 hours, and more preferably between 2 and 24 hours. Rapid release formulations may be achieved by several different methodologies, which may be used alone or in combination.
For example, rapid release may be achieved by a dosage form of sildenafil comprising a rapidly dispersing wafer containing sildenafil or a pharmaceutically acceptable salt thereof which is placed on the tongue and dissolves in the mouth, for example within the buccal fluids. Suitably the wafer is dispersed and/or dissolved over a period of about 1 to 60 sec- onds, preferably about 1 to 30 seconds, most preferably about 1 to 10 seconds. Suitably the wafer is made from a freeze-dried compact containing sildenafil or a pharmaceutically acceptable salt thereof, in a matrix of a buccal fluid-dispersible polymer such as gelatine and a polysaccharide such as mannitol. Sildenafil is dissolved or dispersed into a suspension of mannitol and gelatine prior to filling into blister cavities. These liquid filled blisters are then conveyed through a liquid nitrogen freezing tunnel for freezing and then into a freeze dryer where the solvent is removed leaving behind a highly porous wafer loaded with sildenafil. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61-72 (2000), M J Rathbone, J Hadgraft & M S Roberts in Modified Release Drug Delivery Sys- tems, Marcel Dekker, New York, 2003, US Patent No. 4,642,903 and US Patent No
5,738,875 which are incorporated herein by reference.
Alternatively, rapid release of sildenafil may be provided by the blending and compression of sildenafil with water soluble excipients, such as a sugar such as but not limited to mannitol, and an effervescence agent, at low compression forces. The low compression forces lead to the formation of a highly porous tablet which disintegrates rapidly. Rapid disintegration is further aided by the inclusion of the effervescence agent, which in the context of this specification is defined as one or more agents which produce carbon dioxide upon contact with buccal, gastric, or intestinal fluids.
Typically, effervescence is derived by the reaction which takes place between alkali metal carbonates or bicarbonates and organic acids such as citric acid or tartaric acid to release carbon dioxide. Effervescence may also result from the inclusion of a carbonate or bicarbonate alone to react with acidic gastrointestinal fluids. Suitably the porous tablet disperses over a period of about 1 to 60 seconds, preferably about 1 to 45 seconds, most preferably about 1 to 30 seconds. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61-72 (2000), M J Rathbone, J Hadgraft & M S Roberts in Modified Release
Drug Delivery Systems, Marcel Dekker, New York, 2003, US Patent No. 5,178,878 and US Patent No 5,607,697 which are incorporated herein by reference.
Alternatively, rapid release of sildenafil may be achieved by blending and compressing sildenafil with a suitable sugar such as but not limited to sucrose which has been melt- spun to form a mass of thin filaments with a high surface area. The resulting tablets are highly porous. Upon contact with buccal fluids, they disintegrate rapidly as the mass of thin filaments dissolves. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61-72 (2000) and US Patent No 4,855,326 which are incorporated herein by reference.
Alternatively, rapid release of sildenafil may be achieved by blending and compressing sildenafil with a low mould ability saccharide (e.g. such as but not limited to lactose and mannitol) which has been granulated using a high mould ability saccharide (e.g. such as but not limited to maltose and maltitol) as a binder. The resulting tablets possess characteristics which enable them to dissolve rapidly on contact with aqueous fluids, typically within about 1 to 60 seconds, preferably about 1 to 30 seconds, most preferably about 1 to 15 seconds. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61 -72 (2000) and US Patent No 5,576,014 which are incorporated herein by reference.
Alternatively, rapid release of sildenafil may be achieved by blending and compress- ing sildenafil with a disintegrating agent (e.g. such as but not limited to carboxymethylcellu- lose) and a swelling agent (e.g. such as but not limited to modified starch, e.g. Sodium Starch Glycolate) to produce a rapidly disintegrable tablet which preferably on contact with aqueous fluids disperses over a period of about 1 to 90 seconds, preferably about 1 to 60 seconds, most preferably about 1 to 30 seconds. Details of this technology are described in the scientific and patent literature, for example W Habib et al in Critical Reviews in Therapeutic Drug Carrier Systems, Vol 17 (1) 61-72 (2000) and US Patent No 5,464,632 which are incorporated herein by reference.
It should be appreciated that such tablets will afford advantages over the existing marketed swallow tablets even if swallowed before complete dissolution in the mouth, since dissolution in the gastric fluids will still allow a faster dissolution of sildenafil than is achievable from conventional swallow tablets.
A further embodiment of the present invention relates to the use of taste masking agents and flavours, or the use of forms of sildenafil, for example a suitable salt form, which have a pleasant or acceptable taste.
One way of augmenting the rapid release achievable by a suitable choice of formulation, is to utilise a salt of sildenafil which is very soluble in saliva or in gastric fluid.
Yet another way of augmenting the rapid release achieved by a suitable choice of formulation is to utilise an amorphous form of a salt of sildenafil or sildenafil free base. In addition the amorphous or crystalline form of a salt of sildenafil or sildenafil free base may be dispersed or adsorbed in a thin layer over a high surface area inert substrate. Suitable substrates include but are not limited to: Amberlite ® XAD-4, Amberlite ® XAD-7, Amberlite ® XAD- 16, AMBERSORB ® 348F, AMBERSORB ® 563, AMBERSORB ® 572, Activated carbon, Activated carbon Darco ®, Activated carbon Darco ® G-60, Activated carbon Darco ® KB, Activated carbon Darco ® KB-B, Activated carbon Norit ®, silica gel high purity grades with high pore volume, for example about 0.75 cc/g and average pore diameter 60A.
It will be appreciated that other materials with comparable properties may also be used as substrates.
A solution of sildenafil or a sildenafil salt may be prepared by dissolving the free base or a suitable salt thereof in a suitable solvent or by contacting stoichiometric quantities of the acid and base components of the salt in water or in a solvent such as but not limited to metha- nol, ethanol, or dichloromethane or a mixture thereof, for example at a concentration of 1 % to 20% by weight. The solution is mixed with the inert substrate as defined above, and the required product is isolated by a by either vacuum evaporation or by spray drying. The resulting product has sildenafil or a salt thereof dispersed or adsorbed in a thin layer over a high surface area inert substrate.
The dissolution of sildenafil salts may also be enhanced by reducing the particle size and hence increasing the surface area, for example by such methods as jet milling, ball milling, controlled spray drying, and supercritical fluid precipitation.
Jet or fluid energy milling, involves the use of high pressure air jets to forcibly collide particles together, suitably within a hollow toroidal mill chamber. The high kinetic energy of the air causes particles to impact with other particles with sufficient energy for fracture to occur. This process is repeated until a desired particle size range is obtained. The product is suitably removed from the apparatus with a particle size classifier.
Ball milling of sildenafil salts involves the use of a hollow cylinder rotated along a horizontal longitudinal axis. Inside the cylinder, grinding beads are loaded to a level of 30 to 50% of the chamber volume together with sildenafil salt powder feedstock. The size of the beads depends upon the material being processed and the size of the mill chamber. The chamber is rotated at a suitable velocity to allow the beads to cascade and grind the sildenafil drug particles to a finer and desired size. A feature of ball milling is that it can be conducted in the dry state, alternatively in a wet state in which sildenafil feedstock is supplied to the milling chamber as a suspension in a fluid.
Drug particles of a fine and defined particle size can be manufactured by means of supercritical fluid precipitation. One suitable procedure is rapid expansion of a supercritical solution (RESS), in which sildenafil drug particles are formed as a result of the rapid expansion of a supercritical fluid containing dissolved sildenafil.A second procedure is gas anti-solvent recrystallisation (GAS) in which the supercritical fluid acts as an anti-solvent and causes sildenafil precipitation from a solution. A third procedure is solution enhanced dispersion (SEDS), which involves the rapid dispersion and mixing of a sildenafil solution with the supercritical fluid, typically in a coaxial arrangement, and the extraction of the solvent into the supercritical fluid, leading to precipitation of fine sildenafil drug particles. Multiple repeti- tions of these techniques is also one method for producing novel polymorphic forms which may have greater aqueous solubility and hence be more suitable for rapid release formulations.
Detailed descriptions of both jet and ball milling can be found in Pharmaceutics: the science of dosage forms, M E Aulton (ed) first edition, 2000, Churchill Livingstone, London, D Ganderton in Unit Processes in Pharmacy Volume 7, Heinemann, London, 1968, and M E Fayed & L Often (eds) in Handbook of Powder Science & Technology, second edition, Chapman & Hall, New York, 1997. The procedures described in the above-mentioned references are incorporated herein by reference.
Detailed descriptions of spray drying can be found in K. Masters, Spray Drying in Practice SprayDryConsult International ApS, Denmark, 2002, which is incorporated herein by reference.
Detailed descriptions of supercritical fluid processing can be found in M A McHugh & V J Krukonis, Supercritical Fluid Extraction:Principles & Practice, Butterworth-Heine- mann, Boston, 1994 which is incorporated herein by reference.
Preferably, ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 5 microns. More preferably ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 3 microns. Even more preferably ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 1 micron. Most preferably ultrafine drug particles of sildenafil will have a size profile such that at least 90% by weight of the particles have a maximum diameter no greater than 0.5 microns.
Dispersants and/or other physical stabilisers may be added to prevent aggregation of ultrafine particles and the consequent reduction in the rate of dissolution. Descriptions of suitable dispersants and stabilisers and methods for their use can be found in Dissolution technology, L J Leeson and J T Carstensen (eds), APS, Washington 1974, and C.G. Liversedge et
al., Int J Pharm Vol 125, pages 309-313, 1995), which publications are incorporated herein by reference.
Useful stabilisers include polymers and surfactants e.g. hypromellose, hydroxypro- pylcellulose, mannitol, gelatine, tragacanth, acacia, sorbitan esters, glycerol monostearate, methylcellulose, carboxymethylcellulose, sodium lauryl sulphate, polyvinylpyrrolidone, poly- oxyethylene sorbitan fatty acid esters, polyoxyethylene alkly ethers, polyethylene glycols, block co-polymers of ethylene oxide and propylene oxide.
Delayed release of sildenafil can be achieved by means of a physical barrier or coating which delays exposure of the active material to the buccal, gastric, or intestinal fluids.
One technique which provides delayed release involves the application of a coating of a fluid resistant barrier to a single dosage unit, or to a multiparticulate dosage unit, for example one composed of beadlets, pellets, spheroids, minitablets and/or granules. These coatings can be designed to dissolve at a specific pH range, for example an enteric coating which dissolves at a pH greater than 5.0. Typical pH-dependent polymers suitable for coating dosage forms (single or multiparticulate) include the following:
cellulose acetate phthalate, which dissolves at pH 6.0-6.4
hydroxypropylmethylcellulose phthalate 50, which dissolves at about pH 4.8
hydroxypropylmethylcellulose phthalate 55, which dissolves at about pH 5.2
polyvinylacetate phthalate, which dissolves at about pH 5.0
methacrylic acid-methyl methacrylate copolymer (1 : 1), which dissolves at about pH 6.0 methacrylic acid-methyl methacrylate copolymer (2: 1), which dissolves at pH 6.5-7.5 methacrylic acid-ethyl acrylate copolymer (2: 1), which dissolves at about pH 5.5
hydroxypropylmethylcellulose acetate succinate, which dissolves at about pH 7.0
poly(methylvinylether/maleic acid) monoethylester, which dissolves at pH 4.5 -5.0 poly(methylvinylether/maleic acid)n-butyl ester, which dissolves at about pH 5.4
shellac, which dissolves at about pH 7.0
Alternatively a non-pH-dependant coating may be used, which initially impedes the ingress of aqueous fluid, but subsequently erodes and/or dissolves to expose the active agent to dissolution. Typical non-pH-dependent polymers suitable for coating dosage forms (single or multiparticulate) to provide a fluid resistant barrier which subsequently erodes or dissolves include, but are not restricted to acacia, alginate, amylase, beeswax, carboxymethylcellulose, carnuba wax, cellulose acetate, cholesterol, ethylcellulose, fatty acids, gelatine, glyceryl be- henate, glyceryl monostearate, glyceryl monodistearate, glyceryl tripalmitate, hypromellose, hydroxypropylcellulose, hydrogenated vegetable oil, lecithin, methylcellulose, paraffin wax, pectin, polyethylene glycol, polycapro lactone, polyglycolic acid, polylactic acid, polygly- clide-co-lactide co-polymers, polyvinylprroylidone, starch, stearic acid, stearyl alcohol, partially hydrogenated cottonseed oil/soyabean oil (melting at 51 -55°C), partially hydrogenated
palm oil (melting at 58-63°C), partially hydrogenated cottonseed oil (melting at 61 -65°C), partially hydrogenated soyabean oil (melting at 67-71°C), partially hydrogenated castor oil (melting at 85-88°C), polyethylene glycol 3350 (melting at 54-58°C).
Delayed release of sildenafil may also be achieved by a fluid resistant barrier which combines one or more pH-dependant polymers optionally with one or more non-pH-depend- ant polymers.
Examples of delayed release dosage forms include enteric coated tablets or enteric coated multiparticulate formulations, in which drug-loaded multi-particulate spheres are coated with methacrylic acid-methyl methacrylate co-polymers such as Eudragit LI 00-55, Eudragit L30D-55, or Eudragit FS 30D or Eudragit S100/S12.5. Such formulations will not release sildenafil in the acidic environment of the stomach but only on exposure to the higher pH typically found in the small and large intestine (pH range 5 to 8). An enteric coated tablet illustrating one aspect of this invention may be a single-layer tablet or a multi-layer tablet, such as a bi- or tri-layer tablet, wherein the active agent is present in one or more discrete lay- ers within the compressed tablet form. The discrete tablet layers can be arranged to provide modified or non-modified release of active agent. General descriptions and methods for the preparation of suitable tablets may be found in Aqueous polymeric coatings for pharmaceutical dosage forms, J W McGinty (ed), Marcel Dekker, 1989, New York, and in in Microencapsulation and related drug processes, P Deasy, Marcel Dekker, 1984, New York, which publi- cations are incorporated herein by reference.
Similarly, a capsule can be prepared in which the active dose is provided in the form of sildenafil beads and is divided into two or more parts, each part having a non-pH-depend- ant protective coat of different thickness, which takes a different time to erode. Suitable non- pH-dependent coating materials have already been described above. Further information can be found in J R Robinson & V H Lee (eds) in Controlled Drug Delivery, second edition, Marcel Dekker, New York, 1987 , V Ranade & M A Hollinger in Drug Delivery Systems, second edition, CRC Press, Boca Raton, 2004 and M J Rathbone, J Hadgraft & M S Roberts in Modified Release Drug Delivery Systems, Marcel Dekker, New York, 2003 which are incorporated herein by reference.
Modified release may also be provided in the form of prolonged release. A prolonged release dosage form may consist of a matrix dosage unit, such as a hydrophilic and/or an erodible matrix, usually in tablet form. Release from such a unit can be controlled by a number of mechanisms, such as dissolution, erosion, diffusion, osmotic pressure or any combination thereof. Embodiment of prolonged release dosage forms may utilise excipients which control sildenafil release by more than one formal mechanism.
An erosion controlled prolonged release dosage unit can be achieved by compressing sildenafil with a slowly dissolvable and/or erodable polymeric material into a tablet form.
Release of sildenafil occurs as the polymer dissolves and/or erodes away. Suitable polymers include but are not restricted to glyceryl monostearate, acrylic resins, ethylcellulose, stearyl alcohol, hydroxypropylcellulose, carboxymethylcellulose, hypromellose, methylcellulose, hy- droxyethylmethylcellulose, sodium carboxymethylcellulose. Further information can be found in Controlled Drug Delivery, second edition, J R Robinson & V H Lee (editors), Marcel Dekker, New York, 1987, in Drug Delivery Systems, second edition, V Ranade & M A Hollinger, CRC Press, Boca Raton, 2004, and in Modified Release Drug Delivery Systems, M J Rathbone, J Hadgraft & M S Roberts, Marcel Dekker, New York, 2003 which publications are incorporated herein by reference.
A diffusion controlled prolonged release dosage form may be produced by compressing a water-swellable hydrophilic polymer in combination with sildenafil drug substance. Such systems are often referred to as "hydrophilic matrices" or "swellable-soluble" systems. Water continues to penetrate the matrix causing the swelling of the hydrophilic polymer. The gelatinous layer that is formed, retards the rate of ingress of water into the matrix and the flux of drug out of the matrix. Sildenafil is released from such matrices either by diffusion through the gel layer or by erosion and/or dissolution of the gel layer. Suitable materials would include any pharmaceutically acceptable excipient which can swell and form a gelatinous mass upon hydration, for example, hydroxypropylmethylcellulose, and xanthan gum. Further information and descriptions of such dosage forms can be found in Controlled Drug Delivery, sec- ond edition, J R Robinson & V H Lee (editors), Marcel Dekker, New York, 1987 which publication is incorporated herein by reference.
An osmosis controlled prolonged release dosage form may be produced by compressing sildenafil in combination with an osmagent into a tablet matrix core formulation. This matrix core is then in part coated with a semi-permeable membrane in known manner, utilis- ing such polymers such as methacrylates, ethylcellulose, and cellulose acetate. Aqueous fluids are drawn by osmosis from the exterior environment across the membrane at a controlled rate into the core, causing dissolution of both sildenafil and the osmogent and increased pressure within the matrix core. The pressure forces the solubilised sildenafil out through a specially created aperture or passageway. Examples of osmagents include but are not restricted to sodium chloride, potassium chloride, lithium chloride, magnesium chloride, magnesium sulphate, lithium sulphate, sodium sulphate, potassium sulphate, citric acid, mannitol, ribose, arabinose, galactose, leucine, glycine, fructose, sucrose, sodium and other bicarbonates. Further information can be found in the scientific and patent literature, for example: Controlled Drug Delivery, second edition, J R Robinson & V H Lee (editors), Marcel Dekker, New York, 1987, Modified Release Drug Delivery Systems, M J Rathbone, J Hadgraft & M S
Roberts, Marcel Dekker, New York, 2003, and US Patents 3,760,984, 3,845,770, 3,987,790,
3,916,899, 4008,719, 4,036,227, 4,576,604, 4,578,075, 4,673,405, 4,681,583, 4,693,895, 4,705,515, 4,773, 907, 5,229,133 which documents are incorporated herein by reference.
Prolonged release can also be achieved by applying a porous or semipermeable membrane coat onto a tablet surface by the application of such polymers such as methacrylates, ethylcellulose, and cellulose acetate. Release from such systems can occur by more than one of the mechanisms described above, for example a combination of dissolution, diffusion, erosion, and osmosis. Alternatively, prolonged release can be achieved by coating multiparticulates with semipermeable membranes. The multiparticulates include drug-coated substrates, such as lactose beads, and drug-containing substrates, such as drug-containing lactose spheres.
Hence the compositions of the present invention provide a single modified release formulation of sildenafil or a combination of two or more taken from the various modified and unmodified release forms of sildenafil: rapid release, conventional release, pulsed release, delayed release, and prolonged release. Therefore the compositions of this invention have a component or a combination of components which possess one of the following release characteristics:
Rapid
Delayed
Prolonged
Rapid + Delayed
Rapid + Prolonged
Delayed + Prolonged
Rapid + Delayed + Prolonged
Rapid + Conventional
Delayed + Conventional
Prolonged + Conventional
Rapid + Delayed + Conventional
Rapid + Prolonged + Conventional
Delayed + Prolonged + Conventional
Rapid + Delayed + Prolonged + Conventional
One preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with duration of activity of approximately 2 hours. Such a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conventionally be necessary. Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, and who wishes to avoid distracting or harmful side-effects.
Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided with an initial delay of 0.15 to 2 hours, and with duration of activity of approximately 4 hours. Such a formulation may comprise a conventional release component at a lower dose than would conventionally be necessary, combined with a delayed release component. Such a formulation is useful to a patient who expects a sexual encounter at a time which is not less than 2 hours in the future, but does not wish to be restricted by recent or imminent food intake, and who wishes to avoid distracting or harmful side-effects.
Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with duration of activity of approximately 4 hours. Such a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conventionally be necessary, and a delayed release component. Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, but who is uncertain of the exact timing, and who wishes to avoid distracting or harmful side-effects.
Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided with an initial delay of 0.15 to 2 hours, and with a duration of activity of approximately 12 hours. Such a formulation may comprise a conventional release component at a lower dose than would conventionally be necessary com- bined with either a prolonged release component or a pulsed release component.such a formulation is useful to a patient who expects a sexual encounter at a time which is not less than 2 hours in the future, but is very uncertain of the timing and who does not wish to be restricted by recent or imminent food intake, and who wishes to avoid distracting or harmful side-effects. A b.i.d. presentation of this formulation may be employed for the purposes of continu- ous treatment.
Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with a duration of activity of approximately 12 hours. Such a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conventionally be necessary, and a prolonged release component. Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, and who wishes to be prepared for sexual activity for an extended period of time, and who wishes to avoid distracting or harmful side-effects.
Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided with an initial delay of 0.15 to 2 hours, and with duration of activity of approximately 16-24 hours. Such a formulation may comprise a conventional release component at a lower dose than would conventionally be necessary
combined with either a prolonged release component. Such a formulation is useful to a patient who expects a sexual encounter at a time which is not less than 2 hours in the future, but is very uncertain of the timing and who wishes to be prepared for sexual activity for an extended period of time, and who does not wish to be restricted by recent or imminent food intake, and who wishes to avoid distracting or harmful side-effects.
Another preferred embodiment of this invention is a formulation in which a dose of sildenafil within the therapeutic window is provided both rapidly and with a duration of activity of approximately 16-24 hours. Such a formulation may comprise a rapid release component combined with a conventional release component at a lower dose than would conven- tionally be necessary, and a prolonged release component. Such a formulation is useful to a patient who expects an imminent sexual encounter irrespective of recent food intake, and who wishes to be prepared for sexual activity for a much extended period of time, and who wishes to avoid distracting or harmful side-effects. It should be appreciated that this approach to providing a variety of specially designed formulations to address specific circumstances is es- pecially useful for over-the-counter products where the patient is able to self-diagnose and choose the most appropriate product for his specific needs.
Moreover, it is also believed that these formulations may provide effective therapy for female patients suffering from sexual disorders including but not limited to lack of clitoral arousal. This is especially surprising since the innovator company have reported lack of effi- cacy for sildenafil in female patients.
It should be appreciated that the dose of sildenafil, calculated as the free base, will be adjusted in consideration of the amount of sildenafil in each component and the total amount so as to ensure that blood plasma levels remain within the therapeutic window whilst achieving the effect desired and described above.
The quantity of sildenafil required in each component of each formulation can be determined by the skilled worker from the information provided in this invention. Firstly the target pharmacokinetic profile for the formulation is selected in line with the objects of the present invention. Then, from knowledge of the therapeutic window as defined herein, the mean rate of elimination of sildenafil in the body, and the release profile of sildenafil from each component, it is a matter of routine experimentation to establish the necessary quantity of sildenafil in each component.
In view of the variability of individual patient response to sildenafil, it is envisaged that each of the formulations of this invention will be provided in a range of strengths to permit titration of dose for individual patients.
Preferably, the compositions are in unit dosage form. Unit dosage forms for oral administration may be in tablet or capsule form and may as necessary contain conventional ex- cipients such as binding agents, fillers, lubricants, glidants, disintegrants, effervescent agents, and wetting agents.
Examples of binding agents include but are not limited to: acacia, alginic acid, car- boxymethylcellulose calcium, carboxymethylcellulose sodium, dextrin, dextrose, ethylcellu- lose, gelatin, liquid glucose, guar gum, hydroxyethyl cellulose, hydroxypropyl cellulose, hy- droxypropyl methylcellulose, magnesium aluminium silicate, maltodextrin, methyl cellulose, polymethacrylates, polyvinylpyrrolidone, pregelatinised starch, sodium alginate, sorbitol, starch, syrup, tragacanth.
Examples of fillers include but are not limited to: calcium carbonate, calcium phosphate, calcium sulphate, carboxymethylcellulose calcium, carboxymethylcellulose sodium, compressible sugar, confectioner's sugar, dextrates, dextrin, dextrose, dibasic calcium phosphate dihydrate, dibasic calcium phosphate, fructose, glyceryl palmitostearate, glycine, hydro- genated vegetable oil-type 1 , kaolin, lactose, maize starch, magnesium carbonate, magnesium oxide, maltodextrin, mannitol, microcrystalline cellulose, polymethacrylates, potassium chloride, powdered cellulose, pregelatinised starch, sodium chloride, sorbitol, starch, sucrose, sugar spheres, talc, tribasic calcium phosphate, xylitol.
Examples of lubricants include but are not limited to: calcium stearate, glyceryl monostearate, glyceryl palmitostearate, magnesium stearate, microcrystalline cellulose, sodium benzoate, sodium chloride, sodium lauryl sulphate, stearic acid, sodium stearyl fumarate, talc, zinc stearate.
Examples of glidants include but are not limited to: colloidal silicon dioxide, powdered cellulose, magnesium trisilicate, silicon dioxide, talc.
Examples of disintegrants include but are not limited to: alginic acid, carboxymethylcellulose calcium, carboxymethylcellulose sodium, colloidal silicon dioxide, croscarmellose sodium, crospovidone, guar gum, magnesium aluminium silicate, microcrystalline cellulose, methyl cellulose, polyvinylpyrrolidone, polacrilin potassium, pregelatinised starch, sodium alginate, sodium lauryl sulphate, sodium starch glycolate.
Examples of effervescent agents are effervescent couples such as an organic acid and a metal carbonate or bicarbonate. Suitable organic acids include but are not limited: citric acid, tartaric acid, malic acid, fumaric acid, adipic acid, succinic acid, and alginic acid, and anhydrides and acid salts. Suitable carbonates and bicarbonates include, for example, sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate, magnesium car- bonate, sodium glycine carbonate, L-lysine carbonate and arginine carbonate. Alternatively, only the base component of the effervescent couple may be present.
The solid oral compositions may be prepared by conventional methods of blending, filling or tableting. Repeated blending operations may be used to distribute the active agent throughout those compositions employing large quantities of fillers. Such operations are conventional in the art. The tablets may be coated according to methods known in normal phar- maceutical practice. For example see Pharmaceutical dosage forms: tablets, Volume 1 second edition, H A Lieberman, L Lachman and J B Schwartz (eds) Marcel Dekkker, 1989, New York and G C Cole & J Hogan in Pharmaceutical coating technology, Taylor & Francis, London, 1995 which are herein included by reference.
Formulations of the present invention may be used for the treatment of human male erectile dysfunction, which method comprises administering a formulation of the present invention comprising an effective amount of sildenafil and/or a pharmaceutically acceptable salt thereof to a sufferer in need thereof.
Formulations of the present invention may also be used for the treatment of human female sexual dysfunction, which method comprises administering a formulation of the pre- sent invention comprising an effective amount of sildenafil and/or a pharmaceutically acceptable salt thereof to a sufferer in need thereof.
Formulations of the present invention may also be used in the preparation of a medicament for use in the treatment of human male erectile dysfunction.
Formulations of the present invention may also be used in the preparation of a medic- ament for use in the treatment of human female sexual dysfunction.
The present invention also provides a pharmaceutical composition for use in the treatment of human male erectile dysfunction or human female sexual dysfunction in which the pharmaceutical composition is as defined in the present invention.
The following examples are merely illustrative of the present invention and should not be considered as limiting the scope of the invention in any way.
In Examples 52 to 65 hereinbelow it should be appreciated that the amount of active ingredient is not specified precisely but is readily calculated from a consideration of the target product profile required within the scope of the present invention without undue experimentation. The sildenafil citrate used in these examples may be replaced by other salts of sildenafil with compatible solubility properties.
The present invention also relates to novel compounds and to their use in medical therapy, in particular to their use in the treatment and/or prophylaxis of disorders associated with PDE5 inhibition.
The present invention also relates to processes for preparing these novel compounds. Sildenafil is generically described in US patent 5,250,534 as a selective cGMP PDE inhibitor useful in the treatment of cardiovascular disorders such as angina, hypertension, heart failure and atherosclerosis.
Sildenafil free base i.e. 5-[2-ethoxy-5-(4-methylpiperazin-l -ylsulfonyl)phenyl]-l- methyl-3- propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, is specifically described in US patent 6,204,383 Bl as an agent with pharmaceutical utility in the treatment of male sexual dysfunction.
Sildenafil citrate is the active ingredient in VIAGRA®, which is approved for use in the treatment of MED. The choice of a form of a pharmaceutical agent having a basic functional group is not a matter of routine. The fact that Pfizer (the originator company) markets the citrate salt could lead one skilled in the art to believe that other forms were less preferred since the citrate salt is an unusual choice of salt to market. The choice of salts of sildenafil other than the prior art citrate salt is not therefore prima facie obvious in view of this technical prejudice and other concerns about the formation and properties of such salts. There are problems and technical hurdles to be overcome when selecting a salt other than citrate such as whether a different salt can exist at all, whether the properties of such a salt would be satisfactory, comparable or better than the prior art citrate salt. Whether a suitable method exists for the preparation of a salt other than the prior art citrate salt is not a matter of routine experimentation.
There is a need to find alternative forms of sildenafil other than the prior art citrate which are pharmaceutically acceptable. Such forms apart from finding use in medical therapy and as useful intermediates are also useful in providing new active ingredients containing the active moiety sildenafil which could form the basis for providing new value-added line extenders in the form of advantageous formulations or new uses.
We have found that salts of sildenafil with certain long chain fatty acids can be prepared and have a pleasant and fully acceptable flavour which permits the use of formulations such as chewable tablets, chewing gum, and oral suspensions. The invention accordingly en- compasses such chewable tablets, chewing gum, and oral suspensions, but also is of value in swallow tablets and other conventional formulations. Accordingly, the present invention provides sildenafil salts with long chain fatty acids.
Whilst the compounds of the present invention are prima facie inventive, they also show unexpected advantages and /or overcome technical prejudice. When hereinafter men- tioned, the issue of whether a particular novel form would have advantageous properties in this area over prior art forms would not be predictable. Therefore an advantage of the present novel forms over the prior art forms would be an unexpected advantage. Examples of unexpected advantages are selected from one or more of the following:
Advantages during manufacture in terms of the timing and cost of production, availa- bility of starting materials, reproducibility, and safety.
Advantages during manufacture in terms of improved yield and purity. The yield is established by comparison of the weights of cost-critical starting material and product making
allowance for molecular weights and purities. Purities are established by hplc, gc or other conventional analytical method by means of a validated procedure and comparison with a reference standard. See for example Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA, ISBN 0-683-306472, page 597.
Advantages during manufacture in terms of improved filterability, for example the avoidance of clogging or blinding of filter cloths, and the need for large or expensive or sophisticated filtration apparatus. Filterability testing procedures are based on the concept of Vmax- Vmax modelling is based on the theory that there is some maximum volume of a fluid which will pass a filter at a given pressure. At that point, the flow across the filter will be zero and therefore the resistance of the pad to flow infinite. On the basis of this model the rate of flow of filtrate is proportional to the driving force and the cross-sectional area of the filter bed. Measurements of flow rates and timing of standardised operations may be used to demonstrate advantage.
Advantages during manufacture in terms of improved washability result from the physical properties of the material and the size, shape and surface properties of any particles may be present, which are not per se predictable. Quantification is possible by measurement of, for example, the volume and cost of solvent, duration of agitation, and number of washes required to achieve a standardised reduction in adherent impurity levels. Another relevant fac- tor which would constitute an advantage is a reduction in the loss of product resulting from washing procedures.
Advantages during manufacture in terms of improved ease of drying also result from the physical properties of the material and the size, shape and surface properties of the material, which are not per se predictable. Quantification is achievable by measurement of the length of time and temperature in a specific drying apparatus to achieve a standardised reduction in the solvent level in a standardised quantity of product. Other relevant factors which may give rise to an advantage include the need for agitation, and the need for or suitability for use in efficient apparatus such as filter driers.
Improvements in the colour of a product are linked in perception and often in reality with purity and quality, so may constitute a valuable advantage. Standard colour tests are described in the major pharmacopoeias, for example the European Pharmacopoeia 4th edition 2001, and United States Pharmacopeia 24rd edition 1999-2003 and for example USP 2000 page 1926. Colour may be defined as the perception or subjective response of an observer to the objective stimulus of radiant energy in the visible spectrum extending over a range 400nm to 700nm in wavelength. Three attributes are commonly used to identify a colour: 1) hue, or the quality by which one colour family is distinguished from another, such as red, yellow, blue, green and intermediate terms; 2) value, or the quality that distinguishes a light colour
from a dark one; and 3) chroma, or the quality that distinguishes a strong colour from a weak one, or the extent to which a colour differs from a grey of the same value. The perception of colour and colour matches is dependent on conditions of viewing and illumination. Determinations should be made using diffuse, uniform illumination under conditions that reduce shad- ows and nonspectral reflectance to a minimum. The surfaces of powders should be smoothed under gentle pressure so that a planar surface free from irregularities is presented. Liquids should be compared in matched colour-comparison tubes, against a white background. If results are found to vary with illumination, those obtained in natural or artificial daylight are to be considered correct. Colours of standards should be as close as possible to those of the test specimens for quantifying colour differences. Instrumental methods for measurement of colour provide more objective data than the subjective viewing of colours by a small number of individuals.
The extent to which a product is associated with chemical impurities arising from earlier stages of synthesis is essentially unpredictable and depends both on the synthetic process, the nature of reagents used in the process, and on the physical, chemical, and surface properties of the product. For example a novel salt, polymorph, or pseudopolymorph will have a different and unpredictable profile of trace impurities than a comparator salt. Quantification may be achieved by measurement and characterisation of impurity profiles, for example by GC-MS or LC-MS analysis and comparison with a reference material. Identification of all im- purities is not essential providing sufficient characterisation is obtained from the analytical methodology, though it is of course desirable.
From the point of view of manufacturing and formulation efficiency high bulk density in a product is generally regarded as an advantage, since it allows for smaller apparatus and more acceptable unit doses. Furthermore, the need for costly grinding and compaction proce- dures can be avoided or at least reduced. The European Pharmacopoeia describes definitions and methods by which bulk density of a powder may be measured. Apart from the inherent density of a material which depends on factors such as crystal structure which is unpredictable, there is also the contribution of interp articulate void volume which is equally unpredictable. The bulk density is determined by measuring the volume of a known mass of powder, which has been passed through a screen, into a graduated cylinder. The tapped density is achieved by mechanically tapping a measuring cylinder containing a powder sample. After observing the initial volume, the cylinder is mechanically tapped, and volume readings are taken until little further volume change is observed.
The ability of a powder to flow efficiently through manufacturing apparatus is a sig- nificant factor affecting the economics of manufacture and will vary according to the form of material. For example different salts, polymorphs, and pseudopolymorphs will have different
inherent flow properties, though isolation procedures will also have an effect. Suitable definitions and methods of measurement are described in standard reference texts, for example European Pharmacopoeia 4th edition 2001, and United States Pharmacopeia 24rd edition 1999- 2003 and Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA. Measurement may be for example in terms of the angle of repose, which may be determined experimentally by a number of methods with slightly differing results. A typical method is to pour the powder in a conical heap on a level, flat surface and measure the included angle with the horizontal.
One of the factors affecting the safety of a manufacturing process and hence the cost of the manufacturing facility is the flammability of a material. Typical measurement procedure include the "Burning Rate Test" or "Fire Train Test" as defined in United States Department of Transportation and United Nations regulations (49 CFR 173 Appendix E and UN Recommendations on the Transport of Dangerous Goods, also EEC Directive 79/831 Annex Part A: Methods for the Determination of Physico-Chemical Properties 3.10 Flammability of Solids. A typical test involves applying a source of ignition to a powder "train" measuring 250 mm x 20 mm x 10 mm and measuring the rate at which the powder burns.
Another unpredictable property of particulate pharmaceutical products which affects safety and hence cost of manufacture is the tendency to produce dusts or fines during processing, which dusts or fines vary in their hazardous nature. This property is associated with the static electrical properties of the material. Standard test methods and protocols exist to quantify these problems. For example BS 5958 part 1 - Code of practice for control of undesirable static electricity, British Standards Institute, 1991 ; VDI Fortschritt-Berichte 2263; ISO 6184/1 ; IEC 1241-2-1 , Electrical apparatus for use in the presence of combustible dust Part 2: Test methods, Section 1 : Methods for determining the minimum ignition temperatures of dust. International Electrotechnical Commission, first edition, 1994-12
Other advantages which differentiate unpredictably between alternative forms of a drug substance may be quantified in terms of improved chemical stability. Standard test methods are described in the major pharmacopoeias and are also referenced on the US Food and Drug Administation Web site. Typically accelerated storage tests are performed by stor- age for a period of 1 year or more at elevated temperature (e.g.40°C) and at standard humidity conditions (e.g. 75% RH), with samples being taken at regular intervals of approximately 1 month and assayed for overall purity, specific impurities, and a general impurity screen.
In addition, chemical interactions between drug substance and typical excipients used for formulation will differ for different forms of a drug substance, making one form advanta- geous in one formulation, though not necessarily advantageous in a different formulation. Examples include the interaction between amine drugs and lactose.
Stability to irradiation, especially visible and ultra-violet light is of increasing importance in pharmaceutical science and represents another area in which alternative forms of a drug substance may have significantly and unpredictably different properties. Testing details, such as light source, flux density, and duration are described in Federal Register Notices Vol- ume 62, Number 95, pages 27115-27122, together with recommendations for analytical methodology and assessment of results.
Pharmaceutical materials with relatively high melting points are generally easier to formulate and are subject to less attrition and clumping during processing. Melting points and glass transition temperatures will differ greatly for different salts, polymorphs, pseudopoly- morphs, or other forms of a drug and are in essence unpredictable. Methods for measuring melting points are well-described in the European Pharmacopoeia 4th edition 2001, and United States Pharmacopeia 24rd edition 1999-2003. Various methods are acceptable but differ in detail, for example the melting point determined by the capillary method is the temperature at which the last solid particle of a compact column of a substance in a tube passes into the liquid phase. Suitable apparatus is described in the above mentioned publications and may be calibrated using melting point reference substances such as those of the World Health Organisation or other appropriate substances.
Some materials have a tendency to change their physical form during storage, which can be a disadvantage in pharmaceutical manufacture. For example materials can settle and compact and lose their ability to low freely. One polymorphic form may wholly or partially convert to another over an uncertain time-frame, or solvates and hydrates may lose their solvent or water, and the resultant change in the physical properties of the drug substance can lead to a formulation with uncertain, unreliable, and unpredictable characteristics. Clearly a polymorphic conversion can only occur from a less stable to a more stable form, so there are advantages associated with thermodynamic stability, and the relative stability of a novel form is a priori unpredictable.
All substances absorb moisture when exposed to different relative humidity environments, but the extent and humidity response and temperature response varies very considerably. The term hygroscopicity describes both the rate and the extent of water uptake. It is well established that hygroscopic products are difficult to handle and hence more expensive to formulate. Hygroscopicity is not a priori predictable, and an alternative salt may well be advantageous in this respect. Apparatus for measurement of moisture contents of samples under controlled humidity conditions is available commercially, e.g. from I Holland Ltd., Nottingham, U.K. Simple measurements may be made by monitoring the appearance and weight of samples exposed to atmospheres of known constant humidity and temperature, as described in, for example, The Merck Index 12th edition, Merck and Co Inc.
An important property of a drug substance is its solubility in water and other solvents. There is a link between solubility and bioavailability in as much as very water-insoluble drugs can only be made bioavailable by very careful formulation. The need for high solubility in water or other parenteral media is self-evident, and in general both high, moderate, or low sol- ubility's can be important for different formulations. Formulations designed for sustained release may benefit from very low aqueous solubility. Apparatus and procedures for the measurement of solubility are described in detail in both the European Pharmacopoeia 4th edition 2001, and United States Pharmacopeia 24rd edition 1999-2003.
Another property that influences the ability of a drug substance to go into solution and which varies among different solid forms of a drug substance is the degree of wetting, which affects the rate of dissolution. Wetting is the ability of liquids to form boundary surfaces with solid materials, and is determined by measuring the contact angle which a liquid forms in contact with a solid. The smaller the contact angle the larger the wetting tendency. Wetting phenomena are described in Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA on pages 278-9. In order for immersion of a solid to occur, the liquid must displace air and spread over the surface of the solid. When liquids cannot spread over a solid surface spontaneously, and therefore the spreading coefficient S is negative, it is said that the solid is not wetted. An important parameter reflecting the degree of wetting is the angle made by the liq- uid with the solid surface at the point of contact. By convention, when wetting is complete, the contact angle is 0°. In non-wetting situations it theoretically can increase to a value of 180°, where a spherical droplet makes contact with solid at only one point.
The ability of different forms of a drug to admix with specific common excipients across the formulation / delivery range of technologies is very important and will differ in a non-predictable manner depending on the specific properties of the drug form. Pharmaceutical excipients are substances, other than the active pharmaceutical ingredient, that are used in the finished dosage form. There are very many widely differing excipients each with particular characteristics which form the basis of many widely differing formulations. Excipients and their properties are described in detail in the pharmaceutical literature, for example in Re- mington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA. They serve many functions, for example they stabilise the drug substance by providing antioxidant, heavy -metal chelating, or light- protection properties. They also may be used to enhance bioavailability and to control the release from dosage forms. For solid dosage forms, they provide suitable properties for dis- pensing the drug substance in accurate dosage units that have reproducible release properties. Diluents provide a flowable bulk, binders hold powders together after wet granulation, lubricants provide punch-releasing properties, and disintegrants help to disperse dosage forms in
the GI tract. There is a risk, which may be avoided by careful selection of the form of the drug, of incompatibilities between drug substance and excipients. Screens to detect drug-ex- cipient incompatibilities have been developed using elevated temperature and added water to accelerate potential interactions in ternary and more complex powder blends (Serajuddin ATM et al Pharm Res 1991 8(suppl): SI 03) and such methods have been shown to be capable of rapidly detecting chemical incompatibilities and giving good correlations with results using powder blends of drug and excipients at elevated temperatures and humidity.
The present invention provides amorphous, crystalline or liquid sildenafil salts with long chain fatty acids.
In the first aspect of this invention, the novel salts of sildenafil with long chain fatty acids may be in the form of an amorphous solid. Such amorphous sildenafil salts with long chain fatty acids may be used as an ingredient for inclusion in a range of formulations such as conventional tablets and capsules, in particular, a chewable tablet or formulated into a chewing gum or suspension or liquid for oral administration, or may be prepared in a form in which the salt is absorbed in a carrier, for example an excipient or a mixture of excipients for tabletting or other formulation, or as a solution in a wax or similar pharmaceutically acceptable polymer, such as PEG or PVA. In one preferred aspect of the invention, the salt is formulated as a component of a sweet or chocolate based confection.
Alternatively, sildenafil salts with long chain fatty acids may be in crystalline form. More than one crystalline form may be possible and such polymorphs and pseudo polymorphs including hydrates and solvates also form an aspect of this invention.
Alternatively, sildenafil salts with long chain fatty acids may be in liquid form. Such liquids may be prepared by conventional methods such as dissolving a crystalline or amorphous material in a suitable solvent.
Sildenafil is the active moiety in sildenafil citrate which is the active ingredient in VI¬
AGRA®.
The term fatty acid is understood by one skilled in the art and comprises a monobasic carboxylic acid with a carbon and hydrogen containing substituent group. Long chain is to be understood as comprising a substituent group with six or more carbon atoms. Typically the carbon and hydrogen containing substituent group is a C6-C24 alkyl group or a C6-C24 alkenyl group, i.e. the long chain fatty acid is of formula RC(0)OH wherein R is C6-C24 alkyl or C6- C24 alkenyl.
Examples of salted versions of sildenafil with long chain fatty acids include:
Amorphous sildenafil decanoic acid salt
Amorphous sildenafil docosanoic acid salt
Amorphous sildenafil eicosanoic acid salt
Amorphous sildenafil heneicosanoic acid salt
Amorphous sildenafil heptadecanoic acid salt
Amorphous sildenafil lauric acid salt
Amorphous sildenafil myristic acid salt
Amorphous sildenafil nonadecanoic acid salt
Amorphous sildenafil nonanoic acid salt
Amorphous sildenafil octanoic acid salt
Amorphous sildenafil palmitic acid salt
Amorphous sildenafil pentadecanoic acid salt
Amorphous sildenafil stearic acid salt
Amorphous sildenafil tetracosanoic acid salt
Amorphous sildenafil tricosanoic acid salt
Amorphous sildenafil tridecanoic acid salt
Amorphous sildenafil undecanoic acid salt
Amorphous sildenafil undecylenic acid salt
Crystalline sildenafil decanoic acid salt
Crystalline sildenafil docosanoic acid salt
Crystalline sildenafil eicosanoic acid salt
Crystalline sildenafil heneicosanoic acid salt
Crystalline sildenafil heptadecanoic acid salt
Crystalline sildenafil lauric acid salt
Crystalline sildenafil myristic acid salt
Crystalline sildenafil nonadecanoic acid salt
Crystalline sildenafil nonanoic acid salt
Crystalline sildenafil octanoic acid salt
Crystalline sildenafil palmitic acid salt
Crystalline sildenafil pentadecanoic acid salt
Crystalline sildenafil stearic acid salt
Crystalline sildenafil tetracosanoic acid salt
Crystalline sildenafil tricosanoic acid salt
Crystalline sildenafil tridecanoic acid salt
Crystalline sildenafil undecanoic acid salt
Crystalline sildenafil undecylenic acid salt
The preparation of solid sildenafil free base is described in US patent 6,204,383 Bl . Alternatively, a solution of sildenafil free base may be prepared from the commer- cially available citrate salt by basifying a suspension of the citrate salt in a mixture of water and dichloromethane to a pH of between 8 and 10 and separating the organic phase. Sildenafil base is obtained by evaporation of the organic phase.
The long-chain fatty acids of this invention are available commercially from chemical suppliers such as Aldrich Chemical Company in the UK.
A solution of a salt of sildenafil with a long-chain fatty acid may be prepared by contacting stoichiometric quantities of the acid and base components of the salt, for example by heating an equivalent of sildenafil base and the long chain fatty acid in hot absolute ethanol or methanol at a concentration of 1% to 20% by weight. Alternatively a solution in dichloro- methane may be prepared by heating an equivalent of sildenafil base and the long chain fatty acid in dichloromethane at a similar range of concentrations. Alternatively a mixture of these solvents or other solvents such as may be used. Optionally a miscible co-solvent may be used at a proportion of between 1 :10 and 10: 1. Suitable acids include decanoic acid, docosanoic acid, eicosanoic acid, heneicosanoic acid, heptadecanoic acid, lauric acid, myristic acid, non- adecanoic acid, nonanoic acid, octanoic acid, palmitic acid, pentadecanoic acid, stearic acid, tetracosanoic acid, tricosanoic acid, tridecanoic acid, undecanoic acid, undecylenic acid. Suitable co-solvents include n-propanol, propan-2-ol, acetone, 2-butanone, diethyl ether, toluene, and acetonitrile or a mixture thereof. Amorphous sildenafil long chain fatty acid salts are then isolated by either rapid vacuum evaporation, or spray drying.
If a vacuum evaporation technique is used, it should be carried out as rapidly as possible and under conditions which avoid the presence of seeds of the crystalline salt, to avoid crystallisation of the salt.
If a spray drying technique is used, a concentration of between 2% and 40% weight/volume salt in solution, optionally at elevated temperature, may be used though a concentration of between 10 and 25% is preferred. Aqueous mixtures containing organic solvents may be spray dried in a closed loop spray dryer. If a closed loop spray drier is used the organic solvent content may be raised further up to 100%. The apparatus parameters are ad- justed to give an acceptable product by routine means, but control of outlet gas temperature and solvent content of the outlet gas is particularly important. Hence it is preferred that the outlet temperature is kept above 40°C but below 70°C, more preferably below 50°C, and the solvent content of the outlet gases is kept below 2 grammes per 100 grammes, more preferably below 1.2 grammes per 100 grammes. The technology of spray drying is described in the following publications which are incorporated herein by reference: Spray-drying handbook 5th edition by K Masters, Longman Scientific & Technical, 1991 ISBN 0582062667; Spray- drying of pharmaceuticals for controlled release pulmonary drug delivery by Noha Patel, University of Bath, 2000; Spray-drying of particles intended for inhalation by Kristina Mos'em, Department of Pharmaceutics, Copenhagen, Danish University of Pharmaceutical Sciences 2003, ISBN 8778345243.
In a variation of the spray drying procedure, the solution of sildenafil salt with a long chain fatty acid may be mixed with a suspension of one or more finely powdered excipients or
excipients in solution before spray drying, thus preparing in one step a platform formulation for tabletting or other preparation of a drug product. Known techniques may be employed to coat the particles with enteric or other known coatings for control of drug release after administration.
Drying to the full extent that is desirable for a stable pharmaceutical product is not always practicable during efficient use of the isolation apparatus, particularly in the case of spray drying, so in all the above procedures, a final air or vacuum drying step may be necessary to reduce residual water and solvent to an acceptable level.
A solution of a salt of sildenafil may also be prepared by passing a solution of a strong acid salt of sildenafil or sildenafil in a solvent in which the fatty acid sildenafil salt is soluble through a strong base ion-exchange column in which the column material is in the desired fatty acid salt form. The properties and techniques for conditioning and use of ion exchange resins are generally described in the literature, for example: Ion Exchange Resins 2nd edition by Robert Kunin, John Wiley and Sons, Inc. New York, 1958; The use of ion-ex - change resins as potential bioadhesive drug delivery systems, by Sarah J Jackson, University of Nottingham, UK 1999; Ion exchange resins by Robert Kunin, R.E. Krieger, Malabar, Fla 1990, ISBN 0894644289; BDH Chemicals Ltd: Ion exchange resins 6th edition, BDH Chemicals Ltd, Poole, UK 1981; Ion-exchange resins - properties and applications, Rohm & Haas, Philadelphia; Chromatography edited by E. Heftmann,Van Nostrand Reinhold Company, New York (1975), and these publications are incorporated herein by reference. Suitable ion exchange resins include: AMBERJET™ 4200(C1), Amberlite ® IRA-400 (CI), Amberlite ® IRA-410, Amberlite ® IRA-900, Amberlite ® IRA-743, Dowex ® 1X2-100, Dowex ® 1X2- 200, Dowex ® 1X2-400, Dowex ® 1X4-50, Dowex ® 1X4-100, Dowex ® 1X4-200, Dowex ® 1X4-400, Dowex ® 1X8-50, Dowex ® 1X8-100, Dowex ® 1X8-200, Dowex ® 1X8-400, Dowex ® 21K CI, Dowex ® 2X8-100, Dowex ® 2X8-200, Dowex ® 2X8-400, Dowex ® 22 CI, Dowex ® MARATHON ® A, Dowex ® MARATHON ® A2, Dowex ® MSA-1, Dowex ® MSA-2, Dowex ® 550A OH, Sephadex ® QAE A-25, Sephadex ® QAE A-50, DOW XYS-40013.00®
Crystalline salts of sildenafil with long chain fatty acids are prepared by trituration of the amorphous salts with one of the solvents (or a mixture of solvents) preferably from the following list of preferred solvents, i.e., diethyl ether, di-isopropyl ether, teri-butyl- methylether, di-n-butyl ether, butylvinylether, teri-butylvinylether, tetrahydrofuran, 1,4-diox- ane, n-heptane, n-hexane, cyclohexane, cyclopentane, toluene, o-xylene, m-xylene, p-xylene, dichloromethane, chloroform, carbon tetrachloride, chlorodibromofluoromethane.
Alternatively, a solvent (or a mixture of solvents) from the following list may be used to triturate and crystallise amorphous sildenafil salts with long chain fatty acids: methyl ace-
tate, ethyl acetate, propyl acetate, ethyl formate, isobutyl acetate, isobutyl formate, acetoni- trile, isobutyronitrile, acetone, butanone, isopropylmethylketone, isobutylmethylketone, tert- butylmethylketone, sec-butylmethylketone, w-butylmethylketone, cyclopentanone, cyclohexa- none.
Other more polar solvents which also have utility for trituration are the following: water, methanol, ethanol, n-propanol, propan-2-ol, 1-butanol, isobutyl alcohol, cyclopentanol, 2-ethoxyethanol, 2-methyl-2-butanol, ethyleneglycol, teri-butanol, acetic acid, propionic acid. And, though less preferred, the solvents below may also be used for trituration: N,N-dime- thylformamide, Ν,Ν'-dimethylacetamide, N-methylpyrrolidone, formamide, anisole, sul- folane, nitromethane, Ν,Ν'-dimethylpropyleneurea, dimethylsulfoxide, benzene , chloroben- zene, 1 ,2-dichlorobenzene, 4-methylmorpholine, Ν,Ν,Ν',Ν'-tetramethylethylenediamine, pyridine, diisopropylamine, triethylamine, aqueous ammonia.
Suitable trituration techniques include addition of a small amount of solvent to the amorphous salt, insufficient to cause complete solution, and scratching with a glass rod and/or ultrasonication. Scratching is carried out in a glass tube for periods of approximately 5-10 minutes at a time, then leaving the tube to stand for 1 to 2 hours. Preferably the suspension/paste is brought onto the sides of the vessel during scratching to allow partial evaporation of the solvent. The procedure is then repeated several times. Alternatively the process may be automated by agitating the suspension/paste very vigorously with a small magnetic stirrer in the presence of some quartz anti-bumping granules or other abrasive material such as carborundum. Alternatively the mixture may be subjected to thermal shock by repeated cyclic ultra-freezing with liquid nitrogen followed by heating. Combinations of these techniques may be used.
Another technique which may be employed is to raise the temperature of the amor- phous salt above the glass point and to raise and lower the temperature in a cyclic manner. This technique may be used in combination with thermal shock treatment and scratching.
After trituration, the crystalline product is dried under moderate vacuum to remove excess solvent but not such vigorous conditions as to desolvate any solvate that may have been formed.
The existence of a crystalline salt may be demonstrated by a combination of techniques. Solution nmr and or elemental analysis is used to demonstrate that both acid and base components are present. The salt is characterised by one or more of the following techniques: infra-red spectroscopy, Raman spectroscopy, X-ray powder or single crystal diffraction, solid- state nmr, melting point, DSC, DTA, optical and electron microscopy. The salt may be pre- sent as a hydrate or a solvate.
Once a crystalline salt has been prepared on the small scale by one of the techniques described above so that seeds are available and a suitable solvent has been identified, other
methods may be employed which are more suitable for large scale synthesis. For example, direct crystallisation from a solvent or solvent mixture which has been prepared by mixing the acid and base components optionally at elevated temperature. The solution is made supersaturated by cooling, partial evaporation, or addition of an antisolvent. Alternatively, crystallisa- tion from a supercritical fluid may be employed. Alternatively a salt may be prepared by adding a soluble conjugate salt to a soluble salt of sildenafil in a solvent in which the target salt is insoluble, i.e. the salt of sildenafil with X is prepared by mixing sildenafil/Y with Z/X (a soluble salt of X). For example a salt of sildenafil with a long chain fatty acid may be prepared by adding an ammonium salt of a long chain fatty acid to a solution of a salt of sildenafil with a weak acid such as acetic acid or similar, preferably in the presence of seeds of the crystalline target salt.
Repeated application of the above techniques for inducing crystallisation of amorphous sildenafil salts may give rise to more than one crystal form of each salt, including polymorphs and pseudopolymorphs such as hydrates and solvates. Even when products are the same crystal form as determined by spectroscopic methods, it is often the case that the product that crystallises from one set of solvent conditions will have superior properties from a product obtained from another set of conditions. This may be the result of an improved crystal shape (habit) or density. Additional ways of preparing polymorphs and pseudopolymorphs include repeated small-scale precipitation of salts from a range of solvents (for example the lists of solvents recommended for trituration procedures above) by heating to dissolution, then cooling or partly evaporating. In addition an antisolvent may be added (selected from those solvents which experimentally are found not to dissolve the salt, even with heating). Preferably non-crystalline sildenafil salts are used for these preparations since this minimises the presence of pre-existing crystal forms which may inhibit the production of alternative crystal- line products. Alternatively, hydrates and solvates may be treated with solvents or subjected to a range of different humidities to give other solvates or hydrates. Alternatively, the solvates or hydrates may be subjected to vacuum or oven drying/desolvation, or plunged into an immiscible hot solvent (for example xylene).
Alternatively existing crystalline forms may be subjected to a combination of very high pressure and optionally high temperature, for example melts, or amorphous material maintained above the glass temperature can be induced to crystallise to give a new product. More stable polymorphs may be generated by means of a "polymorph amplifier". This procedure comprises the preparation of a stirred suspension of a crystalline salt in a selected solvent (approximately 5-10% in solution at the lower temperature) and raising the temperature until approximately 90-95% of the solid is dissolved, then allowing the suspension to cool slowly, with stirring, until most of the salt has crystallised. This procedure is repeated cyclically many times, and the product at each stage tested for any change in form.
Crystallisation of novel crystal forms may be brought about by crystallising (for example by evaporation) on quartz or other active surfaces.
Once a novel crystalline form has been produced by application of the above described techniques, improved direct crystallisation techniques can be established by routine experimentation, since seeds of the new form will now be available.
The existence of polymorphs and pseudopolymorphs may be demonstrated by various techniques in combination. Solution NMR and/or elemental analysis is used to demonstrate that both acid and base components are present. The salt is characterised by one or more of the following techniques: infra-red spectroscopy, Raman spectroscopy, X-ray powder or sin- gle crystal diffraction, solid-state NMR, melting point, DSC, DTA, optical and electron microscopy, measurements of density, wetting angle, solubility, stability, and flow properties.
The fatty acid salts of sildenafil of this invention may also be prepared as solid or liquid solutions or dispersions in a liquid or polymeric carrier or matrix.
Such matrix dispersions may be prepared in a variety of ways; the salt may be added to the matrix material either as a solid or in solution and the matrix material itself may also be either in the form of a solid (or liquid, as appropriate) or in solution. If both materials are solids then heating and stirring of a melt may be utilised to form a homogenous mixture before the product is cooled, and either ground to a powder, or left as a liquid or semi-liquid suitable for further formulation. Various techniques are known for the formation of suitable granules and platform products from melts, for example spray congealing techniques to produce pellets have been described by Kanig J.Pharm Sci 53, 188, 1964 and by Kreuschner et al. Acta Pharm. Tech. 23, 159, 1980. A liquid matrix may be used to dissolve the solid salt, or a solution of the matrix product may be formed by mixing a solution of the salt with a solution of the matrix material, and the solvent subsequently removed by evaporation or spray-drying. Suitable solvents for preparing solid or liquid solutions or dispersions include water, common alcohols, ketones, esters and, ethers. Particularly preferred solvents are water, methanol, etha- nol, n-propanol, propan-2-ol, 1-butanol, isobutyl alcohol, cyclopentanol, 2-ethoxyethanol, 2- methyl-2-butanol, ethyleneglycol, teri-butanol, acetone, butanone, isopropylmethylketone, isobutylmethylketone, teri-butylmethylketone, sec-butylmethylketone, ra-butylmethylketone, cyclopentanone, cyclohexanone, methyl acetate, ethyl acetate, propyl acetate, ethyl formate, isobutyl acetate, isobutyl formate, diethyl ether, di-isopropyl ether, teri-butylmethylether, din-butyl ether, butylvinylether, teri-butylvinylether, tetrahydrofuran, 1,4-dioxane, dichloro- methane, acetonitrile, and isobutyronitrile.
In a variation on the procedures described above for the preparation of amorphous salts, the liquid or polymeric carrier or matrix material or solution thereof may form the solvent for the salt formation reaction. Hence the acid and base components may be added separately to the matrix material or solution thereof, optionally with heating to produce a melt or
otherwise ensure a homogenous mixture. The product may then be cooled or evaporated and further treated to produce a form suitable for further formulation.
Suitable ratios of the salt to liquid or polymeric carrier or matrix material may vary from 1 :100 to 10: 1, preferably from 1 :20 to 3:1.
If a volatile solvent is used to form the matrix dispersion, it may be difficult to remove it all by evaporation. In the case of solvents such as water or ethanol this is not a problem and substantial residues may be tolerated, indeed may improve the stability and properties of the product. However residues which decrease the viscosity to the extent that crystallisation may occur on storage are undesirable. Less desirable solvents must be removed suffi- ciently by extended, optionally elevated temperature evaporation to ensure a pharmaceutically acceptable product.
Suitable liquid or polymeric carrier or matrix materials include the following: animal, vegetable or mineral oils, fats, waxes, chocolate, chewing gum base, maize oil, lecithin, groundnut oil, sunflower oil, cottonseed oil, lauroylmonoglyceride, lanolin, gelatin, isinglass, agar, carnauba wax, beeswax, polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), polyethylene glycol esters, ovalbumin, soybean proteins, gum arabic, starch, modified starch, cro- spovidone, hydroxypropyl cellulose, methyl cellulose, carboxymethyl cellulose, sodium car- boxymethyl cellulose, cellulose acetate phthalate, cellulose acetate butyrate, hydroxyethyl cellulose, hydroxypropylmethyl cellulose, ethyl cellulose, chicle, polypropylene, dextrans, in- eluding dexran 40, dextran 70 and dextran 75, dextrins, alpha-, beta-, gamma-cyclodextrins, hydroxypropyl-beta-cyclodextrins, alkylpolyglucosides, chitosan, polyvinylacetate, ethylene vinyl acetate, lectins, carbopols, silicon elastomers, polyacrylic polymers, maltodextrins, lactose, fructose, inositol, trehalose, maltose, raffinose, lauryl alcohol, polysorbate 80, and mixtures.
Preferred materials are PVP and PEG, which are available in various grades differing chiefly in their mean molecular weight. In the case of PVP this may be between 2,000 and 3,000,000, however material in the range 8,000 to 500,000 is more preferred e.g. PVP K-15, K-30, K-60, K-90. In the case of PEG products the mean molecular weight may be in the range 200 to 20,000, but 1,000 to 10,000 is more preferred e.g. PEG 2000, PEG 8000.
It should be appreciated that the salts of the present invention may be prepared on any suitable scale according to the procedures herein outlined and those procedures which are conventional to one skilled in the art of pharmaceutical chemistry, in particular in the preparation of salt forms. Techniques for scale-up are described in the literature for example Pharmaceutical Process Scale-Up by Michael Levin, Marcel Dekker, New York 2003, ISBN 0824706250, which publication is incorporated herein by reference.
Once prepared as described above, amorphous, crystalline and liquid solutions of sildenafil salts with long chain fatty acids may be formulated into pharmaceutical compositions, according to procedures well known in the art. Suitable procedures include those provided in Remington: The Science and Practice of Pharmacy 20th edition, Alfonso R Gennaro editor, Lippincott, Williams, and Wilkins, Philadelphia USA, ISBN 0-683-306472; The art, science, and technology of pharmaceutical compounding by Loyd V Allen, American Pharmaceutical Association, 2001, ISBN 1582120358 - incorporated herein by reference. Suitably, these compositions are adapted for oral use such as tablets, capsules, zydis, gums, candies, chocolates, sachet and oral liquids, or are adapted for topical use such as gels, lotions, patches, or ointments, or are adapted for parenteral use such as intravenous, intramuscular, or subcutaneous injection, or are adapted for use as suppositories, or finally are adapted for inhalation therapy such as bronchial or nasal inhalation therapy.
The invention also provides a co-crystal, which co-crystal comprises sildenafil and a co-crystal former, which co-crystal former is a compound which comprises a phenol moiety. Compounds which comprise a phenol moiety include phenol itself and phenol substituted with one or more (typically from one to three) ring substituents.
Typically, in the co-crystal of the invention, the co-crystal former is a compound of formula (I)

n is an integer of from 1 to 3; and
R is H, -C(0)OR2, -L-C(0)OR2, Ar or -L-Ar;
R2 is H or unsubstituted C alkyl;
L is -CH=CH-
Ar is phenyl or naphthyl, which phenyl or naphthyl is unsubstituted or substituted with from 1 to 3 groups selected from OH and C alkyl; or
Ar is a group of formula (II)

R3 is H or OH, and
m is 0, 1 or 2.
Usually, when R is H, n is 2 or 3.
Often, R2 is H or methyl. Typically, Ar is phenyl, which phenyl is unsubstituted or substituted with from 1 to 3 OH groups; or Ar is typically said group of formula (II) wherein R3 is OH.
The co-crystal former may for instance be selected from compounds having the fol

In one embodiment, the co-crystal former is selected from compounds having the following structures:

and the molar ratio of the sildenafil to the co-crystal former in the co-crystal is 1 : 1.
In another embodiment, the co-crystal former is a compound having the following structure:

and the molar ratio of the sildenafil to the co-crystal former in the co-crystal is 3:2.
The sildenafil in the co-crystal is generally sildenafil base as opposed to a salt of sildenafil, i.e. it is generally 5- {2-ethoxy-5-[(4-methylpiperazin-l-yl)sulfonyl]phenyl}-l- me- thyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one.
The co-crystal of the invention may for instance be any of the co-crystals exemplified herein below. It may be any one of the co-crystals P42-I-A, P42-I-B, P42-III, P42-VI-B, P42-
VI-C, P42-VI-D, P42-VI-E, P42-VII, P42-VIII or P42-IX. It may have the characterisation data of any one of these exemplified co-crystals.
The invention also provides a pharmaceutical composition comprising: (i) a co-crystal of the invention as defined herein, and (ii) a pharmaceutically acceptable carrier.
The following examples are merely illustrative of the present invention and are not intended to limit it in any way:
EXAMPLES EXAMPLE 1
Preparation of amorphous sildenafil decanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichlormethane/methanol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of decanoic acid (0.182 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil decanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.68 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 59.4%; H, 7.8%; N, 13.0%
EXAMPLE 2
Preparation of amorphous sildenafil decanoic acid salt
A solution of sildenafil decanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and decanoic acid (1.82 g) in absolute ethanol (100 ml).
The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phos- phoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 2.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material, .Analysis: C, 59.4%; H, 7.8%; N, 13.0%
EXAMPLE 3
Preparation of crystalline sildenafil decanoic acid salt
Amorphous sildenafil decanoate (approx. 0.02 g) is triturated (as described in the main speci- fication) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 59.4%; H, 7.8%; N, 13.0%
EXAMPLE 4
Preparation of amorphous sildenafil docosanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication. A solution of docosanoic acid (0.36 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil decanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.8 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 64.8%; H, 9.2%; N, 10.3%
EXAMPLE 5
Preparation of amorphous sildenafil docosanoic acid salt
A solution of sildenafil docosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and docosanoic acid (3.6 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 42-46°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 64.8%; H, 9.2%; N, 10.3%
EXAMPLE 6
Preparation of crystalline sildenafil docosanoic acid salt
Amorphous sildenafil docosannoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder dif- fraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta.
Analysis: C, 64.8%; H, 9.2%; N, 10.3%
EXAMPLE 7
Preparation of amorphous sildenafil eicosanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of eicosanoic acid (0.33 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil eicosanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.8 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 64.1%; H, 9.0%; N, 10.7% EXAMPLE 8
Preparation of amorphous sildenafil eicosanoic acid salt
A solution of sildenafil eicosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and eicosanoic acid (3.3 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 43-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 64.1%; H, 9.0%; N, 10.7%
EXAMPLE 9
Preparation of crystalline sildenafil eicosanoic acid salt
Amorphous sildenafil eicosanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 64.1%; H, 9.0%; N, 10.7%
EXAMPLE 10
Preparation of amorphous sildenafil heneicosanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of heneicosanoic acid (0.34 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil heneicosanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.8 g. X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material. Analysis: C, 64.5%; H, 9.1%; N, 10.5%
EXAMPLE 11
Preparation of amorphous sildenafil heneicosanoic acid salt
A solution of sildenafil heneicosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and heneicosanoic acid (3.4 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting: 90°C
Outlet temperature: 44-49°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 64.5%; H, 9.1%; N, 10.5%
EXAMPLE 12
Preparation of crystalline sildenafil heneicosanoic acid salt
Amorphous sildenafil heneicosanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta.
Analysis: C, 64.5%; H, 9.1%; N, 10.5%
EXAMPLE 13
Preparation of amorphous sildenafil heptadecanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of heptadecanoic acid (0.29 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil heptadecanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.79 g. X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material,
Analysis: C, 62.9%; H, 8.7%; N, 11.3% EXAMPLE 14
Preparation of amorphous sildenafil heptadecanoic acid salt
A solution of sildenafil heptadecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and heptadecanoic acid (2.9 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by
Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 62.9%; H, 8.7%; N, 11.3%
EXAMPLE 15
Preparation of crystalline sildenafil heptadecanoic acid salt
Amorphous sildenafil heptadecanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder dif- fraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 62.9%; H, 8.7%; N, 11.3%
EXAMPLE 16
Preparation of amorphous sildenafil lauric acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of lauric acid (0.21 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil laurate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.71 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 60.5%; H, 8.1%; N, 12.5% EXAMPLE 17
Preparation of amorphous sildenafil lauric acid salt
A solution of sildenafil laurate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and lauric acid (2.1 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 60.5%; H, 8.1%; N, 12.5%
EXAMPLE 18
Preparation of crystalline sildenafil lauric acid salt
Amorphous sildenafil laurate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 60.5%; H, 8.1%; N, 12.5%
EXAMPLE 19
Preparation of amorphous sildenafil myristic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of myristic acid (0.24 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil myristate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.74 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 61.5%; H, 8.3%; N, 12.0%
EXAMPLE 20
Preparation of amorphous sildenafil myristic acid salt
A solution of sildenafil myristate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and myristic acid (2.4 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting: 90°C
Outlet temperature: 42-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.5 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 61.5%; H, 8.3%; N, 12.0%
EXAMPLE 21
Preparation of crystalline sildenafil myristic acid salt
Amorphous sildenafil myristate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 61.5%; H, 8.3%; N, 12.0%
EXAMPLE 22
Preparation of amorphous sildenafil nonadecanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of nonadecanoic acid (0.32 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil nonadecanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.82 g. X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material. Analysis: C, 63.7%; H, 8.9%; N, 10.9%
EXAMPLE 23
Preparation of amorphous sildenafil nonadecanoic acid salt
A solution of sildenafil nonandecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and nonadecanoic acid (3.2 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by
Niro.
Inlet temperature setting: 90°C
Outlet temperature: 45-49°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.5 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 63.7%; H, 8.9%; N, 10.9%
EXAMPLE 24
Preparation of crystalline sildenafil nonadecanoic acid salt
Amorphous sildenafil nonadecanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 63.7%; H, 8.9%; N, 10.9%
EXAMPLE 25
Preparation of amorphous sildenafil nonanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication. A solution of nonanoic acid (0.17 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly. The solid amorphous sildenafil nonanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.67 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 58.8%; H, 7.7%; N, 13.3%
EXAMPLE 26
Preparation of amorphous sildenafil nonanoic acid salt
A solution of sildenafil nonanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and nonanoic acid (1.7 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 2.5 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 58.8%; H, 7.7%; N, 13.3% EXAMPLE 27
Preparation of crystalline sildenafil nonanoic acid salt
Amorphous sildenafil nonanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 58.8%; H, 7.7%; N, 13.3%
EXAMPLE 28
Preparation of amorphous sildenafil octanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of octanoic acid (0.152 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly. The solid amorphous sildenafil octanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.65 g. X-ray pow- der diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 58.2%; H, 7.5%; N, 13.6%
EXAMPLE 29
Preparation of amorphous sildenafil octanoic acid salt
A solution of sildenafil octanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and octanoic acid (1.52 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 42-46°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 2.5 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 58.2%; H, 7.5%; N, 13.6% EXAMPLE 30
Preparation of crystalline sildenafil octanoic acid salt
Amorphous sildenafil octanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 58.2%; H, 7.5%; N, 13.6%
EXAMPLE 31
Preparation of amorphous sildenafil palmitic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication. A solution of palmitic acid (0.27 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly. The solid amorphous sildenafil palmitate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.77 g. X-ray pow- der diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 62.4%; H, 8.6%; N, 11.5%
EXAMPLE 32
Preparation of amorphous sildenafil palmitic acid salt
A solution of sildenafil palmitate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and palmitic acid (2.7 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.0 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 62.4%; H, 8.6%; N, 11.5% EXAMPLE 33
Preparation of crystalline sildenafil palmitic acid salt
Amorphous sildenafil palmitate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 62.4%; H, 8.6%; N, 11.5%
EXAMPLE 34
Preparation of amorphous sildenafil pentadecanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of pentadecanoic acid (0.26 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil pentadecanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.76 g. X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material. Analysis: C, 62.0%; H, 8.4%; N, 11.7%
EXAMPLE 35
Preparation of amorphous sildenafil pentadecanoic acid salt
A solution of sildenafil pentadecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and pentadecanoic acid (2.6 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by
Niro.
Inlet temperature setting: 90°C
Outlet temperature: 45-49°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 2.8 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 62.0%; H, 8.4%; N, 11.7%
EXAMPLE 36
Preparation of crystalline sildenafil pentadecanoic acid salt
Amorphous sildenafil pentadecanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 62.0%; H, 8.4%; N, 11.7%
EXAMPLE 37
Preparation of amorphous sildenafil stearic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of stearic acid (0.3 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40 °C and rotated rapidly. The solid amorphous sildenafil stearate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.8 g. X-ray pow- der diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 63.3%; H, 8.8%; N, 11.1%
EXAMPLE 38
Preparation of amorphous sildenafil stearic acid salt
A solution of sildenafil stearate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and stearic acid (3.0 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-47°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.6 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 63.3%; H, 8.8%; N, 11.1% EXAMPLE 39
Preparation of crystalline sildenafil stearic acid salt
Amorphous sildenafil stearate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 63.3%; H, 8.8%; N, 11.1%
EXAMPLE 40
Preparation of amorphous sildenafil tetracosanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication. A solution of tetracosanoic acid (0.39 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil tetracosanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.89 g. X-ray powder diffraction shows a single very broad diffraction peak typical of noncrystalline material. Analysis: C, 65.5%; H, 9.3%; N, 10.0%
EXAMPLE 41
Preparation of amorphous sildenafil tetracosanoic acid salt
A solution of sildenafil tetracosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and tetracosanoic acid (3.9 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.6 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 65.5%; H, 9.3%; N, 10.0% EXAMPLE 42
Preparation of crystalline sildenafil tetracosanoic acid salt
Amorphous sildenafil tetracosanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 65.5%; H, 9.3%; N, 10.0%
EXAMPLE 43
Preparation of amorphous sildenafil tricosanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication. A solution of tricosanoic acid (0.374 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil tricosanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.87 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 65.2%; H, 9.2%; N, 10.1%
EXAMPLE 44
Preparation of amorphous sildenafil tricosanoic acid salt
A solution of sildenafil tricosanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and tricosanoic acid (3.74 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 45-49°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 3.5 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 65.2%; H, 9.2%; N, 10.1% EXAMPLE 45
Preparation of crystalline sildenafil tricosanoic acid salt
Amorphous sildenafil tricosanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 65.2%; H, 9.2%; N, 10.1%
EXAMPLE 46
Preparation of amorphous sildenafil tridecanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultrasonication. A solution of tridecanoic acid (0.226 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil tridecanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.72 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 61.0%; H, 8.2%; N, 12.2%
EXAMPLE 47
Preparation of amorphous sildenafil tridecanoic acid salt
A solution of sildenafil tridecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and tridecanoic acid (2.26 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting
Outlet temperature:
Nozzle diameter
Pump speed (peristaltic)
Feed rate
Nitrogen flow
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 2.8 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 61.0%; H, 8.2%; N, 12.2% EXAMPLE 48
Preparation of crystalline sildenafil tridecanoic acid salt
Amorphous sildenafil tridecanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder diffraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta. Analysis: C, 61.0%; H, 8.2%; N, 12.2%
EXAMPLE 49
Preparation of amorphous sildenafil undecanoic acid salt
Solid sildenafil free base (0.5 grammes) is dissolved in a mixture of dichloromethane/metha- nol (5 ml, 9: 1) with gentle warming and ultras onication. A solution of undecanoic acid (0.2 g) in absolute ethanol is mixed very rapidly in a rotary evaporator flask. High vacuum is applied and the flask is maintained at an external temperature of 40°C and rotated rapidly. The solid amorphous sildenafil undecanoate is isolated as a glass. After evaporation in the rotary evaporator, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 0.7 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 60.0%; H, 7.9%; N, 12.7%
EXAMPLE 50
Preparation of amorphous sildenafil undecanoic acid salt
A solution of sildenafil undecanoate is prepared by heating and ultrasonicating a suspension of sildenafil free base (5.0 grammes) and undecanoic acid (2.0 g) in absolute ethanol (100 ml). The solution is kept hot by means of a jacketed feeder system and spray dried in a commercial closed-loop spray-drying apparatus, Fielder Mobile Minor ® manufactured by Niro.
Inlet temperature setting 90°C
Outlet temperature: 44-48°C
Nozzle diameter 2 mm
Pump speed (peristaltic): 26-28 rpm
Feed rate 2 kg/hour
Nitrogen flow 87 kg/hour
After completion of spray-drying, the flask is placed in a desiccator containing a dish of phosphoric oxide drying agent, and dried under high vacuum to a residual solvent level of below 0.2%. Yield 2.4 g. X-ray powder diffraction shows a single very broad diffraction peak typical of non-crystalline material. Analysis: C, 60.0%; H, 7.9%; N, 12.7% EXAMPLE 51
Preparation of crystalline sildenafil undecanoic acid salt
Amorphous sildenafil undecanoate (approx. 0.02 g) is triturated (as described in the main specification) until the form of the solid is observed to change. Analysis by x-ray powder fraction shows characteristic sharp reflections in the range 5 to 30 degrees 2 theta.
Analysis: C, 60.0%; H, 7.9%; N, 12.7%
EXAMPLE 52: osmotic pump core containing sildenafil citrate
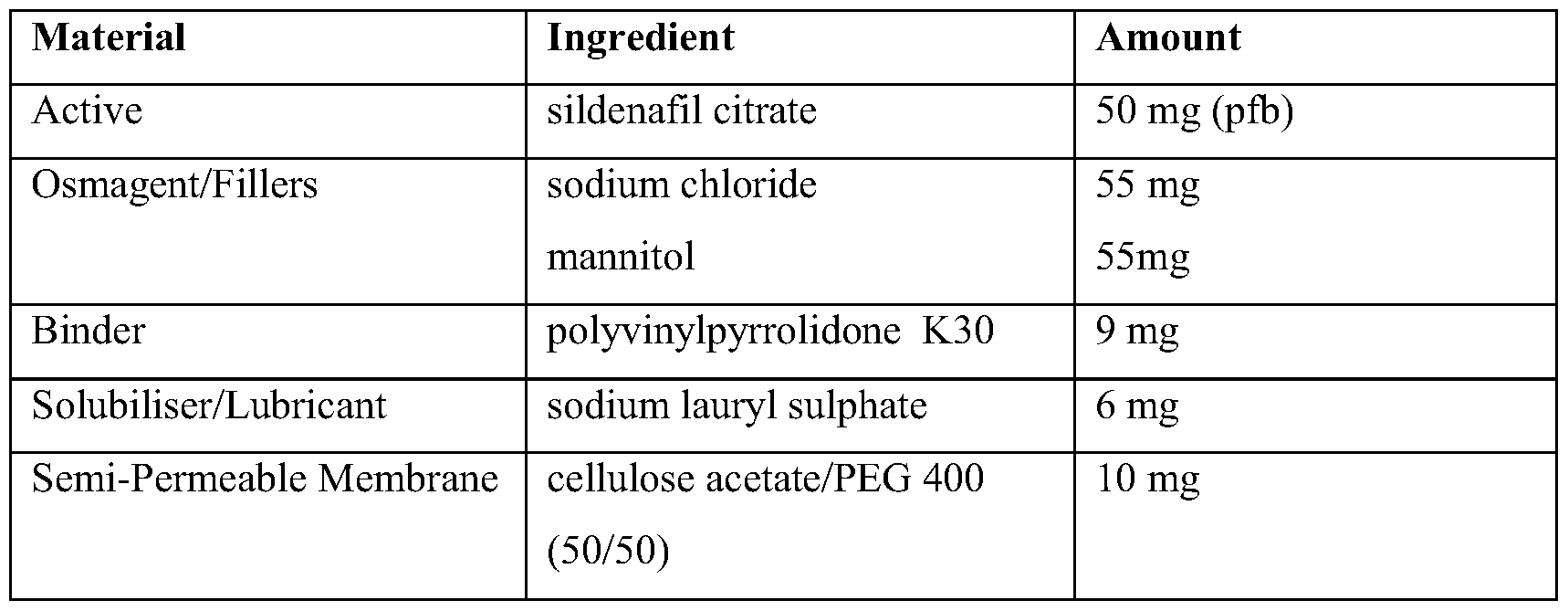
All the sildenafil citrate, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture. The mixture is agitated for 20 minutes or until satisfactory granulation has been achieved. The homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator. After drying, the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release require- ments.
EXAMPLE 53: osmotic pump core containing sildenafil free base
 Active sildenafil 50 mg
Active sildenafil 50 mg
Osmagent/Fillers citric acid 50 mg
sodium chloride 60 mg
Binder polyvinylpyrrolidone K30 9 mg
Solubiliser/Lubricant sodium lauryl sulphate 6 mg
Semi-Permeable Membrane cellulose acetate/PEG 400 10 mg
(50/50)
All the sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid con- sisting of water is sprayed onto the mixture. The mixture is agitated for 20 minutes or until satisfactory granulation has been achieved. The homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator. After drying, the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements. EXAMPLE 54: osmotic pump core containing amorphous sildenafil


All the sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture. The mixture is agitated for 20 minutes or until satisfactory granulation has been achieved. The homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator. After drying,
the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
EXAMPLE 56: osmotic pump core containing ultrafine sildenafil

The ultrafine sildenafil, sodium chloride, the remaining mannitol, polyvinylpyrrolidone, and part of the sodium lauryl sulphate are passed through a 60 mesh screen and mixed in a V- blender for 30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture. The mixture is agitated for 20 minutes or until satisfactory granulation has been achieved. The homogenously
blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator. After drying, the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements. EXAMPLE 57: rapid release layer containing sildenafil mesylate compressed around an osmotic pump core containing sildenafil citrate
Osmotic pump inner core formula

Rapid release outer layer formula

All the sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate for the core are passed through a 60 mesh screen and mixed in a V-blender for
30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture. The mixture is agitated for 20 minutes or until satisfactory granulation has been achieved. The homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator. After drying, the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyeth- ylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typi- cal aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
The rapid release layer is prepared by blending the ingredients from the rapid release formula table. Sildenafil mesylate, half of the micro crystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in a Fielder granulator for 30 minutes. The mixture is then removed and blended for a further 30 minutes with the remaining dibasic calcium phosphate, sodium croscarmellose, and microcrystalline cellulose using a twin shell V-blender. The magnesium stearate is then added to the mixture and mixed together for 5 minutes.
This material is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tableting machine to form a rapid release layer around the cores.
EXAMPLE 58: rapid release layer containing ultrafine sildenafil compressed around an osmotic pump core containing sildenafil citrate
Osmotic pump inner core formula

Semi-Permeable Membrane cellulose acetate/PEG 400 10 mg
( 50/50)
Rapid release outer layer formula

All the sildenafil, sodium chloride, mannitol, polyvinylpyrrolidone, and half of the sodium lauryl sulphate for the core are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture. The mixture is agitated for 20 minutes or until satisfactory granulation has been achieved. The homogenously blended material is then passed through a 30 mesh screen, and dried in a warm fluidised bed granulator. After drying, the granules are milled and passed through a 20 mesh screen, then the remainder of the sodium lauryl sulphate is added and mixed together for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyeth- ylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture until an approximate 10% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typi- cal aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
The sildenafil mixture for the rapid release layer is prepared by loading sildenafil citrate drug substance into a specially contracted 0.5 metre stainless steel ball milling chamber in which the inner surface has been treated with a Teflon coating. To the chamber is added a selection of Zirconium Oxide (Zircoa Inc.) grinding beads with diameters ranging from 1 mm up to 50
mm to fill approximately 50% of the chamber volume. The mill chamber is then rotated at a speed to allow cascading of mill contents to occur, usually a full rotation every 2 seconds. This milling process is continued for 4 days. After day 4, half of the microcrystalline cellulose is added to chamber and the milling process is continued for a further day. The purpose of the microcrystalline cellulose is to act as a carrier substrate for the cohesive ultrafine particles. The milled active is then blended with the remaining microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose in a Fielder a granulator for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose in a V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and mixed together for 5 minutes. The product is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tablet- ing machine to form the rapid release layer for the drug dosage form.
EXAMPLE 59: rapid release layer containing an effervescent couple of sildenafil citrate compressed around an osmotic pump core containing sildenafil citrate
Osmotic pump inner core formula

Pass all the excipients and the active agent separately through a 60 mesh screen. The osmotic pump cores are prepared by the procedures detailed in the preceding Examples 52 onwards.
The sildenafil mixture for the rapid release layer is prepared by blending the ingredients from the rapid release outer layer formula table. The active, half of the microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in a Fielder granulator for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose, microcrystalline cellulose in a twin shell V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and mixed together for 5 minutes. The product is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tableting machine to form the rapid release layer for the drug dosage form.
EXAMPLE 60: rapid release layer of sildenafil citrate sprayed around an osmotic pump core containing sildenafil citrate
Osmotic inner core formula


Pass all the excipients and the active agent separately through a 60 mesh screen. The osmotic pump cores are prepared by the procedures detailed in the preceding Examples 52 onwards.
The rapid release layer of sildenafil is prepared by loading the osmotic pump cores into a Manesty Accela-Cota rotating coating pan and directly film coating the inner cores with a layer of sildenafil citrate mixture until the desired weight gain is achieved. Coating is performed using an inlet temperature between 60-66°C, an exhaust temperature between 39- 40°C, a bed temperature fixed between 43-46°C, an atomising air-pressure of 1.5 to 2.5 bar, an airflow of 1400-1500 m3/hr (800-900cfm), and a spray rate of 200-300 g/min.
In alternative embodiments of the invention, the sildenafil citrate in the rapid release layer may be replaced by amorphous or ultrafine sildenafil citrate or other salts of sildenafil, for example sildenafil mesylate.
EXAMPLE 61 : capsule containing a rapid release tablet of sildenafil mesylate and an osmotic pump tablet containing sildenafil citrate
Osmotic pump tablet

The osmotic core tablet is prepared from the ingredients in the table by the procedures described in the preceding Examples 52 onwards.
The rapid release tablet is prepared by first blending the ingredients in the rapid release tablet table. Thus, the sildenafil mesylate, half of the microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in a Fielder granulator
for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose, microcrystalline cellulose in a twin shell V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and mixed together for 5 minutes. The resulting mixture is then compressed using a Korsch tablet press. The os- motic tablet and the rapid release tablet are encapsulated together into a hard gelation DB capsule Size AA (Capsugel) using a Zanassi 70 capsule filling machine (IMA).
EXAMPLE 62: osmotic pump containing sildenafil citrate with a time delay

Lubricated granules for proton pump cores are prepared from the ingredients in the table by the procedures described in the preceding Examples 52 onwards. The lubricated granules are transferred to a conventional tablet press (Manesty, Fette, Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Ac- cela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (80:20) mixture is sprayed on to the cores from a acetone/water mixture until an approximately 20% increase in weight gain has been achieved. The coated cores are then allowed to cure to ensure that a well-defined coat with full integrity has been achieved. Next a passage way is created through the semipermeable membrane either by mechanical means or using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes are between 0.05 mm up to 2 mm depending upon drug release requirements.
The result of a thicker coat (-20% by weight) and a higher level of cellulose acetate (80%) than in previous Examples is that a delay will occur before the osmotic pump is activated due to the slower ingress of aqueous fluids through the semi-permeable membrane. Modification of the coat thickness and permeability enables control of delay times and rate of release. Typically delay times may be adjusted from as little as 0.5 hrs to about 8 hours.
EXAMPLE 63: osmotic pump core containing sildenafil citrate with a pH dependent delay

The osmotic pump core is prepared from the ingredients in the table by the procedures de- scribed in the preceding Examples 52 onwards.
A delay is induced onto the normal operation of the osmotic pump core by the application of a pH dependant coating, for example a coating that will dissolve at pH greater than 5.0. Hence, application of an enteric coat confers protection to the tablet core until it reaches the small or large intestine. A suitable enteric coat may consist of Eudragit L30 D-55 (30% aqueous dispersion) 76.8% w/w, triethyl citrate 7.7 % w/w, talc 15.5 % w/w, or alternatively the enteric coat could consist of Eudragit S100/S12.5.
EXAMPLE 64: osmotic pump core containing sildenafil citrate with delayed activation, surrounded by a rapid release layer of sildenafil mesylate
Osmotic pump core with delayed activation

Rapid release outer layer
 Filler 30 mg microcrystalline cellulose
Filler 30 mg microcrystalline cellulose
Filler 15 mg dibasic calcium phosphate dihy- drate
Disintegrant 4.25 mg sodium croscarmellose
Lubricant 0.75 mg magnesium stearate
The delayed activation osmotic pump core is prepared from the ingredients of the table by the procedures described in Example 62.
The rapid release layer is prepared by first blending the ingredients in the rapid release outer layer table. All the sildenafil mesylate, half the microcrystalline cellulose, half the dibasic calcium phosphate, and half the sodium croscarmellose are blended in a Fielder granulator for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, sodium croscarmellose, and microcrystalline cellulose using a twin shell V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and mixed together for 5 minutes. This rapid releasing layer is then compressed around the duly formed osmotic pump cores by the employment of a Kilian RUD press-coating tableting machine.
EXAMPLE 65: osmotic core with an enteric coat containing sildenafil citrate, surrounded by a rapid release layer of sildenafil mesylate
Inner osmotic core with an enteric coat

Rapid release outer layer formula
 Filler 30 mg microcrystalline cellulose
Filler 30 mg microcrystalline cellulose
Filler 15 mg dibasic calcium phosphate dihy- drate
Disintegrant 4.25 mg sodium croscarmellose
Lubricant 0.75 mg magnesium stearate
Final weight of IR layer 75 mg
Pass all the excipients and the active agent separately through a 60 mesh screen. Sildenafil, sodium chloride, mannitol, part of the sodium lauryl sulphate and polyvinylpyrrolidone are passed through a 60 mesh screen and mixed in a V-blender for 30 minutes. The dry blended material is then transferred to a large granulator and a granulating fluid consisting of water is sprayed onto the mixture. The mixture is then agitated for 20 minutes at an appropriate blending speed to ensure an adequate endpoint has been achieved. The homogenously blended material is then passed through a 30 mesh screen, prior to drying in a warm fluidised bed granulator. After drying, the granules are milled and passed through a 20 mesh screen prior to the remainder of the sodium lauryl sulphate being added to the granules and mixed for a further 10 minutes. The lubricated granules are then transferred to a conventional tablet press (Korsch) and compressed into 175 mg, 8 mm round biconvex tablet cores. These cores are then transferred into a Manesty Accela-Cota rotating coating pan and a film coat of cellulose acetate/polyethylene glycol (50:50) mixture is sprayed on to the cores from a acetone/water mixture to approximately 10% increase in weight gain has been achieved. The coated cores are then allowed to cure for an appropriate period time and temperature to ensure that a well- defined coat with full integrity has been achieved (0.1 mm to 0.3 mm thick). Next a passage way is created through the semipermeable membrane either through mechanical means or more likely by using a laser drilling machine (Coherent, Convergent Prima). Typical aperture sizes can lie between 0.0 5 mm up to 2 mm depending upon therapeutic rate requirements.
A lag time can be induced on to the osmotic pump by the application of a pH dependant coatings i.e. dissolve at pH's greater than 5.0. In this example, the application of a enteric coat guarantees that the coat dissolves either in the small or large intestine where the pH lies be- tween pH 5.0 to 7.0 as oppose to dissolving in the stomach where the pH lies below 5.0.
The enteric coat consists of Eudragit L30 D-55 (30% aqueous dispersion) 76.8%w/w, Triethyl Citrate 7.7 %w/w, Talc 15.5 % w/w or alternatively the enteric coat could consist of Eudragit S100/S12.5.
The rapid releasing layer is prepared by first blending the ingredients in the above table. Sildenafil mesylate, half of the microcrystalline cellulose, half of the dibasic calcium phosphate and half of the sodium croscarmellose are blended in fielder granulator for 30 minutes. The mixture is then removed and blended with the remaining dibasic calcium phosphate, so- dium croscarmellose, microcrystalline cellulose using a twin shell V-blender for a further 30 minutes. The magnesium stearate is then added to the mixture and then mixed for 5 minutes. The resulting product is then compressed around the osmotic pump cores by means of a Kilian RUD press-coating tableting machine to form the rapid release layer for the drug dosage form.
The principles described in the above Examples can be applied to the production of sildenafil dosage forms with any combination of rapid, conventional, delayed and prolonged release characteristics to deliver the blood plasma profiles required for the improved formulations of this invention, and to deliver sildenafil within the therapeutic window either more rapidly than the currently marketed swallow tablets containing sildenafil, or continuously for a long period of time compared to the currently marketed swallow tablets containing sildenafil, or more rapidly and continuously for a long period of time when compared to currently marketed swallow tablets containing sildenafil. Pharmacological Data
In the prescribing information for VIAGRA™, published as reference 69-5485-00-9, under the heading of clinical pharmacology, in particular under the sub-heading of "pharmacokinetics and metabolism", mean sildenafil plasma concentrations in healthy male volunteers after administration of a single dose of 100 mg of sildenafil are provided in figure 1. Under the sub-heading "pharmacodynamics" it is stated that "The time course of effect in one study, showing an effect for up to 4 hours but the response was diminished compared to 2 hours." This information is herein incorporated by reference.
The formulations of the present invention are tested in the animal and human models de- scribed in the patents and references herein mentioned and found to be PDE5 inhibitors. The formulations of the present invention can therefore be used in the treatment and/or prophylaxis of the disorders described in the above mentioned patents and are incorporated herein by reference. Guidance is to place the tablet under the tongue for 60 seconds (see caffeine summary where we tested how much caffeine dissolved by doing this as substitute for Viagra) then swallow the tablet. Described herein is our model of how the tablet dissolves based on
different H levels by our estimates we will deliver the equivalent sildenafil in plasma somewhere around to a normal dose of 100 mg with about 112% to 116% of AUC (so within 505b2 guidelines). We will model and/or test in human next to confirm. EXAMPLE 66: Sildenafil Cocrystal Screening
This Example describes the results of the project entitled "Cocrystal screening of P42" conducted at the Scientific and Technological Centers of the University of Barcelona (CCiT-UB). The study has been performed by the technicians of the Polymorphism and Calorimetry Unit (CCiT-UB) in collaboration with the X-ray diffraction Unit (CCiT-UB). The main aim of the study was the search of new cocrystals of Sildenafil (referenced as P42) by performing a co- crystal screening.
1. Bibliographic precedents
Patent EP0463756A1 (1991): it claims preparation methods for synthesis of Sildenafil base and other pyrazolopyrimidinethione derivates. PXRD diffractogram and DSC/TGA thermograms are not reported.
Mustafa M. E 1-Abadelah, Salim S. Sabria, Monther A. Khanfar, Wolfgang Voelte and Cacilia Maichle-Mossmerc. Zeitschrift fur Naturforschung B., (1999) Volume 54b, Pages 1323-1326. A crystal structure is reported: iso-Sildenafil base (CCDC: CAXZEG).
Patent WO02102802A (2002): it claims preparation methods for the synthesis of Sildenafil base and other pyrazolopyrimidinethione derivatives. PXRD diffractogram and DSC/TGA thermograms are not reported.
Hemmige S. Yathirajan, Basavegowda Nagaraj, Padmarajaiah Nagarajaa and Mi- chael Bolteb. Acta Crystallographica, (2005), 61, Pages 489-491. A crystal structure is reported: Sildenafil citrate monohydrate (CCDC: FEDTEO).
Iwona Wawer, Maciej Pisklak and Zdzislaw Chilmonczyk, Journal of Parmaceuti- cal and Biomedical Analysis, (2005), 38, Pages 865-870. Sildenafil base and its citrate lU,l3C and 15N NMR spectra.
- Rahul Banerjee, Prashant M. Bhatt and Gautam R. Desijaru, Crystal Growth & Design, (2006), 6(6), Pages 1468-1478. The following crystal structures are reported: Sildenafil saccharinate hemikis (ethanol) clathrate (CCDC: QEKVEI), Sildenafil saccharinate methanol clathrate (CCDC: QEKVIM), Sildenafil saccharinate hemikis (dimethylsulfoxide) clathrate (CCDC: QEKVOS), Sildenafil saccharinate nitrome- thane clathrate (CCDC: QEKVUY), Sildenafil saccharinate hemikis (pyrrolidinone) clathrate (CCDC: QEKWAF), Sildenafil saccharinate (CCDC: QEKVEJ), Sildenafil
sacchannate formamide clathrate (CCDC: QEKWIN), Sildenafil saccharinate hemikis (1,4-dioxane) clathrate (CCDC: QEKWOT), Sildenafil saccharinate hemikis (ethylene glycol) clathrate (CCDC: QEKWUZ), Sildenafil saccharinate hemikis (dimethylformamide) clathrate (CCDC: QEMLEA), Sildenafil saccharinate acetonitrile clathrate (CCDC: QEMLIE) and Sildenafil saccharinate hemikis dehydrate clathrate (CCDC: QEMLOK).
Mahmoud M. Al Omari, Mohammad B. Zughul, J. Eric D. Davies and Adnan A. Badwan, J. Inch Phenom. Macrocycl. Chem (2007) 57 Pages: 379-384. Sildenafil: β-cyclodextrim complex. PXRD diffractogram and DSC/TGA thermograms are not reported.
Patent WO2007080362A1 (2007): it claims Sildenafil Acetylsalicylic acid cocrys- tals. A PXRD diffractogram, crystal data and DSC/TGA thermograms are reported. Patent WO2007110559A1 (2007): it claims: Sildenafil hydrochloride (polymorphs I, II and III); Sildenafil hydrogensulfate (polymorphs I, II and III); Sildenafil hemi- sulfate (polymorph I); Sildenafil hemitartrate (polymorphs I and II); Sildenafil esyl- ate (polymorphs I, II, III and IV) and Sildenafil fumarate (polymorph I). PXRD dif- fractograms, crystal data (sildenafil esylate polymorph I) and a DSC thermogram are reported.
Patent PT16633364E (2008): preparation methods for the synthesis of Sildenafil base and Sildenafil citrate. PXRD diffractogram and DSC/TGA thermograms are not reported.
Patent WO2010146407A: it claims nanoparticles of Sildenafil citrate. A PXRD diffractogram (Figure 6) is reported.
Patent RU2012101818A: PXRD diffractogram and DSC/TGA thermograms are not reported.
Dmitry s Stepanovs and Anatoly Mishnev, Zeitschrift f r Naturforschung B., (2012) Volume 67, Issue 10, Pages 491-494. Crystal structure is reported: Sildenafil base (form I) (CCDC: QEGTUT).
Palash Sanphui, Sridu Tothadi, Sommath Ganguly and Gautam R. Desijaru, Molecular Pharmaceutics, (2013), 10, Pages 4687-4697. The following crystal structures are reported: Sildenafil hemikis (oxalate) (CCDC: YIWWIM), Sildenafil hydrogen- fumarate trihydrate (CCDC: YIWWOS), Sildenafil hemikis (succinate) monohy- drate (CCDC: YIWWUY), Sildenafil hemikis (glutarate) (CCDC: YIWXAF), Sildenafil adipic acid cocrystal (CCDC: YIWXEJ), Sildenafil pimelic acid cocrystal (CCDC: YIWXIN), Sildenafil suberic acid cocrystal (CCDC: YIWXOT) and Sildenafil sebacic acid cocrystal (CCDC: YIWXUZ).
Miroslav Zegarac, Edislav Leksic, PrimozSket, Janez Plavec, Maja Devcic Bogda- novic, Dejan-Kresimir Bucar, Miljenko Dumic and Ernest Mestrovic,
CrystEngComm (2014), 16, 32-35. A Sildenafil base: Acetylsalicylic acid cocrystal is reported: Crystal data: triclinic, PI, a = 9.6707(7) A, b = 12.3070(7) A, c = 14.5432(12) A, a = 85.266(2)°, β = 74.549(5)°, γ = 82.829(6)°, V = 1653.1(2) A3, T =298(2)K and Z =2.
Somchai Sawatdee, Chaveng Pakawatchai, Kwanjai Nitichai, Teerapol Srichana and Hirihattaya Phetmung. Saudi Pharmaceutical Journal. (2015), 23, Pages 504-514. Crystallographic data of Sildenafil base (CCDC: QEGTUT) and Sildenafil citrate monohydrate (CCDC: FEDTEO) are reported.
Avani P. Khristi, Dr.Tejal Soni and Dr. B.N.Suhagia. Indo American Journal of Pharmaceutical Research, (2015), 5(7), Pages 2700-2708. Sildenafil citrate: Aspirin cocrystal. A PXRD diffractogram and a DSC thermogram are reported.
Dmitrijs Stepanovs, Mara Jureb and Anatoly Mishneva. Mendeleev Commun., (2015), 25, Pages 49-50. Sildenafil salycilate: Crystal data: triclinic, P-l, a = 9.5030(3), b = 11.6242(4) and c = 14.2074(6) A, a = 99.167(1)°, β = 108.370(1)°, γ = 92.229(2)°, V = 1463.75(9) A3, T = 173(2) K and Z = 2. The compound melts between 179-182 °C.
The calculated PXRD diagram from the crystal structure (CCDC) of the published P42 form in comparison to the initial batch of P42 (sildenafil base) as received is shown in figure 1.
2. Results and Discussion
2.1. Characterization of the initial batch
Initially, the sample supplied with reference Sildenafil base, batch: 101071417 (P42) (EXT- 15-108) has been characterized by means of PXRD, DSC, TGA and 'H-NMR. This sample corresponds to the same crystal structure reported in literature (CCDC ref. code: QEGTUT), according to their PXRD diffractograms (Figure 1). The original sample has been analyzed by PXRD after grinding and it remains unchanged (Figure 2). A melting point of 189°C is observed in the DSC analysis and no weight loss is observed in the TGA analysis from room temperature to 300°C.
2.2 Solubility study of P42
Initially, a solubility study of P42 in 30 solvents has been conducted at r.t. and at high temperature. P42 is soluble at r.t. in the following solvents: MeOH, EtOH, formic acid, MEK, acetone, MiBK, DMF, DMSO, AcOEt, THF, DME, dioxane, DCM, chloroform, acetic acid, benzyl alcohol and diethylamine; it is soluble at 50°C in IP A, butanol, ACN, toluene, xylene and
NH3 (32%) in water. It is insoluble in ethylene glycol, H2O, pentane, heptane, cyclohexane, Et20 and DIE.
The solutions obtained from solubility experiments were kept sealed at room temperature for 24 hours and the solids which precipitated were isolated and analysed. In cases where no solid precipitated after 24 hours, the solutions were kept air-opened at room temperature until a solid crystallized. Anhydrous form I of sildenafil base has been obtained in some cases. Moreover, the following new forms have been isolated and characterized by means of DSC, TGA, PXRD and ¾-NMR in some cases:
Form II: is obtained in most of the screening methodologies applied using ACN as solvent. According to its DSC, TGA and 'H-NMR this form could be assigned to a new anhydrous form. DSC analysis shows in some batches an exothermic transitions and in others a melting and crystallization process. Its PXRD diagram has been in- dexed (a=35.82 A, b=17.14 A, c=8.14 A, V=2483 A3).
Form P42-A: is obtained in most of the screening methodologies applied using toluene as solvent. According to its TGA and ¾-NMR this form could be assigned to a hemitoluene solvate. Its PXRD diagram has been indexed (a=15.07 A, b=14.52 A, c=12.85 A, β=106.54°, V=2691 A3).
- Form P42-B: is obtained in the solubility study by using formic acid. According to its TGA and ¾-NMR this form could be assigned to a formic acid salt. It could be attributed to two molecules of formic acid per molecule of P42. Its PXRD could not be indexed.
Form P42-C: is obtained in the solubility study by using dioxane. According to its TGA and 'H-NMR this form could be assigned to a dioxane solvate. It could be attributed to one molecule of dioxane per molecule of P42. Its PXRD diagram has been indexed (a=37.44 A, b=45.95 A, c=11.33 A, α=153.87°, β=40.94°, γ=146.89° V=4658 A3).
Form P42-D: is obtained in the solubility study by using acetic acid. According to its TGA and 'H-NMR this form could be assigned to an acetic acid cocrystal. Its PXRD diagram has been indexed (a=19.20 A, b=14.76 A, c=9.76 A, β=97.90°, V=2737 A3).
Form P42-E: is obtained through slurry experiments by using chloroform as solvent.
According to its TGA and ¾-NMR this form could be assigned to an hydrate. Its
PXRD diagram has been indexed (a=15.07 A, b=14.52 A, c=12.85 A, β=106.54°, V=2691 A3).
A comparison of the PXRD diagrams of these new forms of P42 is shown in Figures 3 and 4. The diagram in Figure 5 summarizes the different behaviour of forms I and II of P42.
2.3. Cocrystal screening
2.3.1. Coformers
The following 13 coformers have been used after considering the virtual cocrystal prediction conducted with P42 (table 1).
Table 1. Coformers used in P42 cocrystal screening
Coformers Description1
OB O Linear formula: C15H10O7
Quercetin
MW: 302.24 g/mol
HO'" ¾i'"'No'" 5:i ..OH
(2)
Melting point: 316°C
Linear formula: C14H12O3
Resveratrol MW: 228.24 g/mol (3) Melting point: 253-255°C
 Melting point: 261-263°C
Melting point: 261-263°C
Phlor

Methyl 3,4,5-trihydroxybenzoate Linear formula: CsHgOs (Methyl gallate) MW: 184.15 g/mol (5)
 Melting point: 201-203°C
Melting point: 201-203°C
Linear formula: C9H8O4
Ca
°C,

pka: 4.62
Linear formula: C4H6O6
OH O
MW: 150.09 g/mol
Tartaric Acid HO,
Melting point: 170-172°C (7)
pKal : 2.99 and pKa2: 4.40
1 According to Sigma Aldrich description.
Linear formula: C7H6O3
3hyeroxy benzoic acid MW: 138.12 g/mol
(8) Melting point: 200-203°C
 pKal : 4.06 and pKa2: 9.92
pKal : 4.06 and pKa2: 9.92
0.v .,OH Linear formula: C7H6O3
MW: 138.12 g/mol
4-Hydroxybenzoic Acid
Melting point: 213-217°C (9) OH pKal : 4.48 and pKa2: 9.32
2-tert-butylbenzene-l,4-diol Linear formula: C10H14O2 (teri-Butylhydroquinone) MW: 166.22 g/mol
(10)
 Melting point: 127-129°C
Melting point: 127-129°C
Re

O^OCCBakCHs Linear formula: C10H12O5
3 4 5 trichyeroxy propoxy ester
MW: 212.20 g/mol (12)
OH Melting point: 146-149°C
Linear formula: C7H6O4
3,4 - Dihydroxybenzoic Acid MW: 154.12 g/mol
(13) Melting point: 197-200°C
 pKal : 4.48 and pKa2: 8.83
pKal : 4.48 and pKa2: 8.83
Linear formula: C6¾02
Sorbic acid MW: 112.13 g/mol (14)
 Melting point: 132-135°C pKa: 4.8
Melting point: 132-135°C pKa: 4.8
2.3.2. Drop-Grinding (DG) experiments
Drop grinding experiments were undertaken with 20-50 mg of the total mixture
(API: coformer, 1 : 1) and one drop of four different solvents. 32 experimental combinations of P42 with 8 solid coformers in 4 solvents were performed (table 2).
Table 2. Drop-grinding experiments with P422
Coformer Solvent
2 Results: (0) negative result, (1) positive result: P42 + coformer + new peaks observed by PXRD
# IPA ACN Toluene THF
2 Quercetin 1 1 1
3 Resveratrol 0 1 0 0
4 Phloroglucinol 1 1 0 0
5 Methyl gallate 1 1 0 1
7 Tartaric Acid 1 1 1 1
8 3-Hydroxybenzoic Acid 1 1 1 1
9 4-Hydroxybenzoic Acid 1 1 1 1
10 teri-Butylhydroquinone 0 0 0 0
2.3.3. Reaction Crystallization (RC) and Slurry (SY) experiments
Reaction crystallization or slurry experiments have been performed with different solvents. Thirteen coformers have been selected according to the results obtained from DG experiments. Reaction Crystallization (RC)
Depending on the solubility, a saturated solution of the most soluble component (P42 or the coformer) in different solvents was prepared in a sealed vial under stirring. A small quantity of the less soluble component was added until it did not dissolve anymore. The suspension was kept under stirring for one day and the resulting solids were filtered and analyzed by PXRD. Slurries (SY)
Suspensions of P42 and different coformers in a 1 : 1 molar ratio (40-80 mg of the final mixture) in selected solvents were prepared. The sealed tubes were kept under stirring for one day and the resulting solids were filtered and analyzed by PXRD (table 3). Table 3. RC or SY of P42 with 13 coformers in different solvents3
Solvent
Coformer
IPA ACN THF
Quercetin RC/2 SY/1 SY/2
Resveratrol RC/0 RC/0 SY/1
Phloroglucinol SY/0 SY/0 SY/0
Methyl gallate RC/1 RC/2 SY/1
Caffeic Acid RC/0 SY/2 SY/1
3 Results: (0) negative, (1) positive: P42 + coformer + new peaks observed in PXRD, (2) positive: cocrystal or salt.
Tartaric Acid RC/1 RC/1 RC/2
3- Hydroxybenzoic Acid RC/1 RC/2 RC/1
4- Hydroxybenzoic Acid RC/2 RC/2 SY/2 teri-Butylhydro quinone SY/0 SY/0 SY/0
Resorcinol SY/0 SY/2 SY/0 Propyl gallate RC/0 SY/0 SY/0 3,4-Dihydroxybenzoic Acid SY/2 RC/1 SY/1 Sorbic Acid RC/0 RC/0 SY/0
2.3.4. Identification and characterization of the new forms
From the cocrystal screening, some new multicomponent phases have been isolated and characterized by means of DSC, TGA, PXRD and 'H-NMR. A brief description of the new forms is described as follows:
- P42-I: P42: Quercetin.
o P42-I-A: It has been obtained by reaction crystallization in IPA. According to its TGA and ¾-NMR analysis, it could be attributed to 1 molecule of quercetin and 1 molecule of IPA per molecule of P42. Its PXRD diagram has been indexed (a=22.43 A, b=15.79 A, c=l 1.24 A, β=92.21°, V=3975 A3) with a number of impurities equal to zero, o P42-I-B: It has been obtained by slurry in THF. According to its TGA and 'H-NMR analysis, it could be attributed to 1 molecule of quercetin and 2A molecules of THF per molecule of P42. Its PXRD diagram has been indexed (a=22.52 A, b=13.53 A, c=8.01 A, α=57.96°, β=95.99°, γ=95.95° V=2047 A3 ) with a number of impurities equal to zero.
P42-III: P42:Methyl gallate. It has been obtained by reaction crystallization in ACN. According to its 'H-NMR, it could be attributed to 1 molecule of methyl gallate per molecule of P42. Its PXRD diagram has been indexed (a=12.98 A, b=13.45 A, c=l 1.99 A, a=l 16.63°, β=82.45°, γ=118.59° V=1628 A3) with a number of impurities equal to zero.
- P42-IV: P42:Tartaric acid.
o P42-IV-A: It has been obtained by drop grinding in IPA. According to its ¾- NMR analysis, it could be attributed to 1 molecule of tartaric acid per molecule of P42. Its PXRD diagram has been indexed (a=18.07 A, b=13.61 A, c=7.59 A, α=85.980, β=92.34°, γ=110.08° V=1741 A3) with a number of impurities equal to zero,
o P42-IV-B: It has been obtained by slurry in THF. According to its 'H-NMR analysis, it could be attributed to 1 molecule of tartaric acid per 2 molecules of P42. Its PXRD diagram has been indexed (a=33.57 A, b=14.05 A, c=11.55 A, V=2483 A3 ) with a number of impurities equal to zero.
P42-V-A: P42:3-Hydroxybenzoic acid. It has been obtained by reaction crystallization in ACN. According to its 'H-NMR analysis, it could be attributed to 1 molecule of 3-hydroxybenzoic acid and ¼ molecule of ACN per molecule of P42. The PXRD diagram could not be indexed.
P42-VI: P42:4-Hydroxybenzoic acid.
o P42-VI-B: It has been obtained by drop grinding in ACN. According to its 'H-NMR analysis, it could be attributed to 1 molecule of 4-hydroxybenzoic acid per molecule of P42. The PXRD diagram could not be indexed, o P42-VI-C: It has been obtained by reaction crystallization in IPA. According to its TGA and ¾-NMR analysis, it could be attributed to 1 molecule of 4- hydroxybenzoic acid and ½ molecule of IPA per molecule of P42. The cell could not be indexed,
o P42-VI-D: It has been obtained by reaction crystallization in ACN. According to its ¾-NMR analysis, it could be attributed to 1 molecule of 4-hydroxybenzoic acid per molecule of P42. The PXRD diagram could not be indexed.
o P42-VI-E: It has been obtained by slurry in IPA. According to its TGA and ¾-NMR analysis, it could be attributed to 1 molecule of 4-hydroxybenzoic acid and ¼ molecule of IPA per molecule of P42. The cell could not be indexed.
P42-VII: P42:Resorcinol. It has been obtained by slurry in IPA. According to its ¾- NMR analysis, it could be attributed to 1 molecule of resorcinol per molecule of P42. The PXRD diagram has been indexed: (a=l 1.27 A, b=14.83 A, c=14.18 A α=38.10ο, β=94.79°, γ=96.08° V=1453 A3) with a number of impurities equal to zero.
P42-VIII: P42:3,4-dihydroxybenzoic acid. It has been obtained by slurry in IPA. According to its 'H-NMR analysis, it could be attributed to 1 molecule of 3,4-dihy- xoxybenzoic acid and 1 molecule of IPA per molecule of P42. The PXRD diagram has been indexed: (a=12.76 A, b=13.46 A, c=12.1 1 A a=l 12.03°, β=84.09°, γ=1 14.28° V=1750 A3) with a number of impurities equal to zero.
P42-IX: P42: Caffeic acid. It has been obtained by slurry in IPA. According to its ¾- NMR analysis, it could be attributed to 2 molecules of caffeic acid and 1 molecule of water per 3 molecules of P42. Its PXRD diagram has been indexed (a=25.84 A, b=8.36 A, c=20.31 A, β=121.19°, V=3744 A3) with a number of impurities equal to zero.
The PXRD of these new multicomponent forms of P42 are shown in figures 6 and 7.
The following table 4 summarizes the pKa values of the different coformers which are interacting with P42 (form I (CCDC: QEGTUT), pKa: 8.44) as a cocrystal or as a salt. The interaction has been studied according to the crystal structures published in CCDC. The acid coformers used in the present screening are included.
Table 4. pKa values of different coformers
CCDC
Coformer pKa values Salt or Cocrystal
code
pKal : 3.1 / pKa2:
Citric Acid FEDTEO Salt monohydrate
4.8
Saccharin pKa: 1.6 QEMLOK Salt dihydrate
pKal : 1.2 / pKa2:
Oxalic Acid YIWWIM Salt
4.2
pKal : 3.0 / pKa2:
Fumaric Acid YIWWOS Salt trihydrate
4.4
pKal : 4.2 / pKa2:
Succninic Acid YIWWUY Salt monohydrate
5.6
pKal : 4.3 / pKa2:
Glutaric Acid YIWXAF Salt
5.4
pKal : 4.4 / pKa2:
Adipic Acid YIWXEJ Cocrystal
5.4
J. Chem. Eng. Data 2009, 54, 2914-2917.
pKal : 4.5 / pKa2:
Pimelic Acid YIWXIN Cocrystal
5.4
pKal : 4.5 / pKa2:
Suberic Acid YIWXOT Cocrystal
5.4
pKal : 4.6 / pKa2:
Sebacic Acid YIWXUZ Cocrystal
5.6
Salicylic Acid pKa: 3.5 Caffeic Acid pKa: 4.6 P42-IX
pKal : 3.0 / pKa2:
Tartaric Acid P42-IV
4.5
3 -Hydroxybenzoic pKal : 4.1 / pKa2:
P42-V
Acid 9.9
4-Hydroxybenzoic pKal : 4.5 / pKa2:
P42-VI
Acid 9.3
3 ,4-Dihydroxyben- pKal : 4.5 / pKa2:
P42-VIII zoic Acid
Sorbic Acid pKa: 4.8
According to these data we can assess the formation of salt or cocrystal depending on the coformer pKa values. Thus, coformers with pKa values higher than 4.5 will probably form a cocrystal, coformers with pKa values lower than 4.2 will probably form a salt, coformers with pKa values between 4.3 and 4.5 will require the crystal structure to confirm/discard the proton transfer. However, the confirmation of salt/cocrystal formation will always need the determination of the crystal structure by means of SXRD. In this sense, we have prepared a battery of crystallizations of all the new forms discovered during this study.
3. Conclusions
A new anhydrous form of P42 has been obtained through a slurry experiment in ACN and it has been characterized.
Five new forms of P42 have been isolated from the solubility study and they have been characterized: P42-A (toluene solvate), P42-B (formic acid salt), P42-C (dioxane solvate), P42-D (acetic acid salt or cocrystal) and P42-E (hydrate).
Several multicomponent forms of P42 have been obtained through a cocrystal screening with nine of the 13 coformers used: quercetin, phoroglucinol, methyl gallate, caffeic
acid, tartaric acid, 3-hydroxybenzoic acid, 4-hydroxybenzoic acid, resorcinol and 3,4- hydroxybenzoic acid.
The new multicomponent form of P42 with Quercetin (1 :1 stoichiometry) has been obtained in two different forms: one IPA solvate form and one THF solvate form, which have been isolated and characterized.
The new multicomponent form of P42 with Phloroglucinol has not been isolated and characterized in pure form.
The new multicomponent form of P42 with Methyl gallate (1 :1 stoichiometry) has been isolated and characterized.
- The new multicomponent form of P42 with Tartaric acid has been obtained in two different forms: (1 : 1 stoichiometry) and (2: 1 stoichiometry), which have been isolated and characterized. According to the pKa values these forms are expected to be salts. The new multicomponent form of P42 with 3 -Hydroxybenzoic acid (1 :1 stoichiometry) has been obtained in ACN solvate form, which have been isolated and characterized. According to the pKa values this form is expected to be a salt.
The new multicomponent form of P42 with 4-Hydroxybenzoic acid (1 : 1 stoichiometry) has been obtained in two different polymorphic forms, one IPA solvate form and one THF solvate form, which have been isolated and characterized. One of these forms has not been isolated and characterized in pure form. According to the pKa values these forms are expected to be cocrystals.
The new multicomponent form of P42 with Resorcinol (1 : 1 stoichiometry) has been isolated and characterized.
The new multicomponent form of P42 with 3,4-Dihydroxybenzoic acid (1 : 1 stoichiometry) has been obtained in IPA solvate form, which have been isolated and charac- terized. According to the pKa values this form is expected to be a cocrystal.
The new multicomponent form of P42 with Caffeic acid (2:3 stoichiometry) has been obtained as a hydrated form, which have been isolated and characterized. According to the pKa values this form is expected to be a cocrystal.
EXAMPLE 67
Sildenafil Citrate Compression Coating

Aerosil 200 (Hydrophilic fumed silica) 1.500 0.500
Magnesium Stearate 3.000 1.000
Total 300.000 100.000
Sustained Release Core
Contains 112.389mg of Sildenafil Citrate. This amount is the equivalent of 80mg Sildenafil base per tablet.
Method of manufacture
• All components are dispensed (except the lubricant) into a Pestle and Mortar
• Mix for 4 minutes
• Dispense the Magnesium Stearate (Lubricant)
• Mix for 1 minute
Compressed on a Manesty F3 compression machine using 9mm Round Normal Convex tooling
• Target weight: 3 OOmg
• Target Hardness: lOkp
Proposed Immediate Release Layers
Two different sizes have been trialled with the aim of deciding the most appropriate size to be carried forward; the outer layer contains 28.097mg of Sildenafil Citrate per tablet. This is equivalent to 20mg of Sildenafil base.


Isomalt 721 811.603 90.178
Sodium Starch Glycolate 36.000 4.000
Peppermint Flavour 501500 TP0504 9.000 1.000
Stevia 1.800 0.200
Sucralose 9.000 1.000
Magnesium Stearate 4.500 0.500
Total 900.000 100.000
Method of Manufacture
• Sildenafil Citrate dispensed and mixed in a Pestle and Mortar using a Geometric dilution method with Isomalt 721
· All other components (except lubricant) dispensed and added to the Pestle and Mortar
• Mix for 2 minutes
• Dispense the Magnesium Stearate (Lubricant)
• Mix for 1 minute
250mg - Compressed on a Manesty F3 compression machine using 9mm Round Normal Con- vex tooling
• Target weight: 250mg
• Target Hardness: 5kp
900mg - Compressed on a Manesty F3 compression machine using 14mm Round Normal Convex tooling
· Target weight: 900mg
• Target Hardness: 5kp
Final proposed manufacture combing the two formulations
• The Sustained release core will be made as described previously (but with flat tooling)
· Depending on the final size required the tooling will be changed to 10 - 20mm Flat tooling
• The tablet die will be filled with approximately 50% of the Immediate release blend
• The Sustained Release core will be placed centrally into the pre-filled die.
• The remaining Immediate release blend will be added
· Tablet will be compressed to a suitable hardness
Dissolution Summary
Aim: To produce dissolution profiles at the following conditions for immediate release/sus- tained release development formulations and the comparator product (Pfizer Viagra tablets).
• 0.01M hydrochloric acid (pH 2.0)
• pH 4.5 phosphate buffer
• pH 6.8 phosphate buffer with CTAB
Analytical Testing
Tablets were tested using dissolution with UV endpoint to obtain drug release profiles.
Dissolution conditions (0.01M hydrochloric acid, pH 2.0)
Apparatus USP I (basket)
Dissolution media 0.01 M hydrochloric acid
Volume dissolution me900ml
dia
Temperature 37°C
Speed lOOrpm
Sampling points As detailed in data for specific sample
Detection UV at 267nm
Dissolution conditions (pH 4.5 buffer)
Apparatus USP I (basket)
Dissolution media 0.13 M potassium monophosphate (KH2PO4) adjusted to pH 4.5 with sodium hydroxide
Volume dissolution me900ml
dia
Temperature 37°C
Speed lOOrpm
Sampling points As detailed in data for specific sample
Detection UV at 267nm
Dissolution conditions (pH 6.8 buffer with surfactant)
Apparatus USP I (basket)
Dissolution media 50 mM sodium phosphate buffer (NaH2P04), 0.125% CTAB, adjusted to pH 6.8
Volume dissolution me- 900ml
dia
Temperature 37°C
Speed lOOrpm
Sampling points As detailed in data for specific sample
Detection UV at 267nm
Tablet details:
Sildenafil (Pfizer) Dose
25mg (as 35.1mg sildenafil citrate)
50mg (as 70.2mg sildenafil citrate)
lOOmg (as 140.5mg sildenafil citrate)
Sustained Release Development Product
16CF25/026
112.39mg sildenafil citrate (equivalent to 80mg
sildenafil base)
Immediate Release Development Product
16CF25/027
28.097mg sildenafil citrate (equivalent to 20mg
sildenafil base)
1. Results for dissolution performed in 0.01M hydrochloric acid 1.1 Viagra 25mg

1.2 Viagra 50mg
50mg Viagra Tablets
% dissolved
Time -point (mins)
Vessel 4 Vessel 5 Vessel 6
0 0 0 0
5 101 98 105
10 101 98 105
15 101 99 106
20 102 99 106
30 102 100 106
45 103 100 107
60 103 100 106 .3 Viagra lOOmg
1 OOmg Viagra Tablets
% dissolved
Time -point (mins)
Vessel 1 Vessel 2 Vessel 3
0 0 0 0
5 93 100 98
10 100 101 99
15 101 102 100
20 101 102 100
30 101 98 101
45 100 102 100
60 101 103 101 .4 Sustained-release development formulation - 16CF25/026
16CF25/026
Time -point (hours ) % dissolved
Vessel 1 Vessel 2 Vessel 3
0 0 0 0
0.5 23 21 16
1 38 35 29
1.5 53 50 43
2 67 63 57
2.5 80 75 70
3 90 87 82
3.5 97 97 92
4 98 101 97
4.5 98 101 97 Immediate-release development formulation - 16CF25/027
16CF25/027 - 0.01 M HC1
% dissolved
Time -point (minutes)
Vessel 1 Vessel 2 Vessel 3
0 0 0 0
5 89 86 83
10 103 104 102
15 103 105 102
Results for dissolution performed in pH 4.5 phosphate buffer 2.1 Viagra 25mg

2.2 Viagra 50mg tablets

5 101 102
10 103 105
15 104 106
20 103 105
30 100 104
45 104 106
60 106 108
2.3 Viagra lOOmg tablets


% dissolved
Time -point (minutes)
Vessel 1 Vessel 2 Vessel 3
0 0 0 0
5 93 86 88
10 101 100 102
15 101 100 102
Results for dissolution performed in pH 6.8 phosphate buffer with 0.125% CTAB
3.1 Viagra 25mg tablets

3.2 Viagra 50mg tablets

20 83 81 82
30 83 86 86
45 88 90 90
60 90 92 92
90 93 95 93
120 94 97 95
150 97 98 96
3.3 Viaera lOOrae tablets
1 OOmg Viagra Tablets
% dissolved
Time -point (mins)
Vessel 1 Vessel 2 Vessel 3
0 0 0 0
5 65 58 59
10 68 64 64
15 73 66 66
20 73 68 67
30 77 71 70
45 78 74 72
60 82 76 75
Note - Dissolution was only run for 60 minutes, not fully dissolved 3.4 Sustained-release development formulation - 16CF25/026


EXAMPLE 68
Sildenafil Citrate Compression Coating

Sustained Release Core
Contains 112.389mg of Sildenafil Citrate. This is the equivalent of 80mg Sildenafil per tablet. Method of manufacture
• All components are dispensed (except the lubricant) into a Pestle and Mortar
• Mix for 4 minutes
• Dispense the Magnesium Stearate (Lubricant)
• Mix for 1 minute
Compressed on a Manesty F3 compression machine using 9mm Round Normal Convex tooling
• Target weight: 300mg
• Target Hardness: lOkp Immediate Release Layer
Formulation Details
An immediate release tablet was formulated with API Sildenafil base (250 mg tablet containing 20mg sildenafil base) with composition shown in the following table.

Method of Manufacture
• Sildenafil base dispensed and mixed in a Pestle and Mortar using a Geometric dilution method with Isomalt 721
• All other components (except lubricant) dispensed and added to the Pestle and Mortar
• Mix for 2 minutes
• Dispense the Magnesium Stearate (Lubricant)
• Mix for 1 minute
250mg - Compressed on a Manesty F3 compression machine using 9mm Round Normal Convex tooling
• Target weight: 250mg
• Target Hardness: 5kp
Final proposed manufacture combing the immediate release formulation of sildenafil base 16CF25/033 with the sustained-release core formulation 16CF25/026 of sildenafil citrate described in Example 67
• The Sustained release core will be made as described previously in Example 67 (with flat tooling)
• Depending on the final size required the tooling will be 10 - 20mm Flat tooling
• The tablet die will be filled with approximately 50% of the Immediate release blend
• The Sustained Release core will be placed centrally into the pre-filled die.
• The remaining Immediate release blend will be added
Tablet will be compressed to a suitable hardness Analytical Testing of Immediate Release Sildenafil Base Formulation 16CF25/033
Tablets of Immediate release formulation 16CF25/033 were tested using a dissolution system with UV endpoint to obtain drug release profiles in pH 4.5 phosphate buffer and 0.01M hydrochloric acid media.
Dissolution conditions:

16CF25/033 in pH 4.5 phosphate buffer
16CF25/033 - pH 4.5
Time -point (min)
Vessel 3 Vessel 4 Mean
0 0 0 0
5 41 34 38
10 70 70 70
15 83 80 82
20 87 86 86
30 90 86 88
45 91 88 90
60 93 89 91
Claims
1. A pharmaceutical composition comprising sildenafil or a pharmaceutically acceptable salt or co-crystal thereof admixed with excipients in a multicomponent pharmaceutical com- position, wherein a first component is adapted to deliver sildenafil rapidly to promote fast onset of action, and a further component is adapted to deliver the sildenafil from dose to dose, wherein the sildenafil is delivered from dose to dose within the therapeutic window.
2. A pharmaceutical composition according to claim 1 comprising said first component which is adapted to deliver sildenafil rapidly to promote fast onset of action, a second component which is adapted to deliver the sildenafil from dose to dose, and a third component wherein the sildenafil is delivered from dose to dose within the therapeutic window.
3. A pharmaceutical composition according to claim 1 or claim 2 wherein the pharma- ceutical composition is a dosage form.
4. A pharmaceutical composition according to any one of claims 1 to 3, comprising: a first component which is adapted to deliver the sildenafil rapidly to promote a fast onset of action, and
a second component which is adapted to deliver the sildenafil from dose to dose, and which is further adapted to also deliver the sildenafil from dose to dose within the therapeutic window.
5. A pharmaceutical composition according to any one of the preceding claims wherein the pharmaceutical composition is in the form of a swallow tablet.
6. A pharmaceutical composition according to claim 5, wherein the tablet comprises: a first component which is adapted to deliver the sildenafil rapidly to promote a fast onset of action, and
a second component which is adapted to deliver the sildenafil from dose to dose, and which is further adapted to also deliver the active ingredient from dose to dose within the therapeutic window.
7. A pharmaceutical composition according to any one of claims 4 to 6, wherein the first component comprises sildenafil admixed with one or more excipients which promote rapid release.
8. A pharmaceutical composition according to any one of claims 4 to 7, wherein the first component comprises sildenafil admixed with one or more excipients which promote immediate release.
9. A pharmaceutical composition according to claim 7 or claim 8 wherein the first component comprises from 10% to 30% by weight of the total amount of sildenafil in the first and second components, optionally wherein the first component comprises 20% by weight of the total amount of sildenafil in the first and second components.
10. A pharmaceutical composition according to any one of claims 7 to 9, wherein the first component comprises from lOmg to 30mg of sildenafil admixed with said excipients, optionally wherein the first component comprises 20mg of sildenafil admixed with said excipients.
11. A pharmaceutical composition according to any one of claims 7 to 9, wherein the first component comprises from 5mg to 15mg of sildenafil admixed with said excipients, optionally wherein the first component comprises lOmg of sildenafil admixed with said excipients.
12. A pharmaceutical composition according to any one of claims 7 to 9, wherein the first component comprises from 2.5 to 7.5 mg of sildenafil admixed with said excipients, option- ally wherein the first component comprises 5mg of sildenafil admixed with said excipients.
13. A pharmaceutical composition according to any one of the preceding claims wherein the first component is a rapidly disintegrating component comprising: said sildenafil, a disintegrating agent and optionally a swelling agent.
14. A pharmaceutical composition according to claim 13 wherein the first component comprises: said sildenafil; a disintegrant which is a dissacharide alcohol, carboxymethylcellu- lose or sodium croscarmellose; and optionally a modified starch.
15. A pharmaceutical composition according to claim 13 or claim 14 wherein the first component comprises said sildenafil, isomalt and sodium starch glycolate.
16. A pharmaceutical composition according to any one of claims 1 to 12 wherein the first component is a rapidly disintegrating component comprising: said sildenafil in a matrix of a buccal fluid-dispersible polymer, and a polysaccharide.
17. A pharmaceutical composition according to claim 16 wherein the buccal fluid-dispersible polymer comprises gelatine, or wherein the polysaccharide comprises mannitol, or wherein the buccal fluid-dispersible polymer comprises gelatine and the polysaccharide comprises mannitol.
18. A pharmaceutical composition according to any one of claims 1 to 12 wherein the first component is a rapidly disintegrating component comprising: (i) said sildenafil, (ii) an effervescence agent, and (iii) a disintegrating agent or a water soluble excipient.
19. A pharmaceutical composition according to claim 18 wherein the effervescence agent comprises an alkali metal carbonate or bicarbonate and optionally an organic acid.
20. A pharmaceutical composition according to claim 18 or claim 19 wherein the water soluble excipient is a sugar, optionally wherein the sugar is mannitol.
21. A pharmaceutical composition according to any one of claims 1 to 12 wherein the first component is a rapidly disintegrating component comprising: said sildenafil and a melt- spun sugar which comprises filaments of the sugar.
22. A pharmaceutical composition according to claim 21 wherein the sugar is sucrose.
23. A pharmaceutical composition according to any one of claims 1 to 12 wherein the first component is a rapidly disintegrating component comprising: said sildenafil, a low mould ability saccharide and a high mould ability saccharide, wherein the low mould ability saccharide is granulated using the high mould ability saccharide as a binder.
24. A pharmaceutical composition according to claim 23 wherein the low mould ability saccharide is lactose or mannitol and/or the high mould ability saccharide is maltose or malt- itol.
25. A pharmaceutical composition according to any one of claims 1 to 12 wherein the first component is a rapidly disintegrating component comprising: said sildenafil which is dispersed or adsorbed over a high surface area inert substrate.
26. A pharmaceutical composition according to claim 25 wherein the substrate is an ion exchange resin, a polymeric absorbent, activated carbon, or silica gel.
27. A pharmaceutical composition according to claim 25 or claim 26 wherein the substrate is selected from: Amberlite ® XAD-4, Amberlite ® XAD-7, Amberlite ® XAD-16, AMBERSORB ® 348F, AMBERSORB ® 563, AMBERSORB ® 572, Activated carbon, Activated carbon Darco ®, Activated carbon Darco ® G-60, Activated carbon Darco ® KB, Ac- tivated carbon Darco ® KB-B, Activated carbon Norit ®, and silica gel.
28. A pharmaceutical composition according to any one of the preceding claims wherein the sildenafil in the first component is a pharmaceutically acceptable salt of sildenafil.
29. A pharmaceutical composition according to any one of the preceding claims wherein the sildenafil in the first component is sildenafil citrate.
30. A pharmaceutical composition according to claim 29 wherein the first component further comprises a taste masking agent or a flavouring agent.
31. A pharmaceutical composition according to any one of claims 1 to 27 wherein the sildenafil in the first component is sildenafil free base.
32. A pharmaceutical composition according to any one of claims 1 to 27 wherein the sildenafil in the first component is in the form of a co-crystal of sildenafil.
33. A pharmaceutical composition according to claim 32 wherein the co-crystal is as defined in any one of claims 95 to 103.
34. A pharmaceutical composition according to any one of claims 1 to 27 wherein the sildenafil in the first component is a salt of sildenafil with a fatty acid.
35. A pharmaceutical composition according to claim 34 wherein the salt of sildenafil with a fatty acid is as defined in any one of claims 105 to 108.
36. A pharmaceutical composition according to any one of claims 4 to 35 wherein the second component comprises sildenafil and one or more excipients which promote modified release.
37. A pharmaceutical composition according to claim 36 wherein the second component comprises from 70% to 90% by weight of the total amount of sildenafil in the first and second
components, optionally wherein the second component comprises 80% by weight of the total amount of sildenafil in the first and second components.
38. A pharmaceutical composition according to any one of claims 4 to 37 wherein the second component comprises from 70mg to 90mg of sildenafil, and one or more excipients which promote modified release, optionally wherein the second component comprises 80mg of sildenafil, and one or more excipients which promote modified release.
39. A pharmaceutical composition according to any one of claims 4 to 37 wherein the second component comprises from 35mg to 45mg of sildenafil, and one or more excipients which promote modified release, optionally wherein the second component comprises 40mg of sildenafil, and one or more excipients which promote modified release.
40. A pharmaceutical composition according to any one of claims 4 to 37 wherein the second component comprises froml7.5mg to 22.5 mg sildenafil, and one or more excipients which promote modified release, optionally wherein the second component comprises 20mg of sildenafil, and one or more excipients which promote modified release.
41. A pharmaceutical composition according to any one of claims 4 to 40 wherein the sildenafil in the second component is sildenafil citrate.
42. A pharmaceutical composition according to any one of claims 4 to 40 wherein the sildenafil in the second component is sildenafil free base.
43. A pharmaceutical composition according to any one of claims 4 to 40 wherein the sildenafil in the second component is a salt of sildenafil with a fatty acid, optionally wherein the salt of sildenafil with a fatty acid is as defined in any one of claims 105 to 108, or the sildenafil in the second component is in the form of a co-crystal of sildenafil, optionally wherein the co-crystal is as defined in any one of claims 95 to 103.
44. A pharmaceutical composition according to any one of claims 36 to 43, wherein the excipients which promote modified release in the second component comprise one or more polymers.
45. A pharmaceutical composition according to any one of claims 36 to 44 wherein the second component comprises: said sildenafil and a hydrophilic matrix suitable for promoting prolonged release of the sildenafil.
46. A pharmaceutical composition according to claim 45 wherein the hydrophilic matrix comprises a polymer which is a cellulose ether or xanthan gum.
47. A pharmaceutical composition according to claim 46 wherein the cellulose ether is selected from carboxymethylcellulose (CMC), methylcellulose (MC) and derivatives thereof, hydroxyethylcellulose (HEC) and derivatives thereof, hydroxylpropyl cellulose (HPC), hy- droxypropylmethylcellulose, and ethylcellulose (EC).
48. A pharmaceutical composition according to any one of claims 45 to 47 wherein the second component further comprises a filler, optionally wherein the filler is lactose.
49. A pharmaceutical composition according to any one of claims 45 to 48 wherein the second component further comprises hydrophilic silica.
50. A pharmaceutical composition according to any one of claims 36 to 44 wherein the second component comprises: said sildenafil and a dissolvable or erodible polymer suitable for promoting prolonged release of the sildenafil.
51. A pharmaceutical composition according to claim 50 wherein the polymer is selected from glyceryl monostearate, acrylic resins, ethylcellulose, stearyl alcohol, hydroxypropyl- cellulose, carboxymethyl-cellulose, hypromellose, methylcellulose, hydroxyethyl-methyl- cellulose, sodium carboxymethylcellulose.
52. A pharmaceutical composition according to any one of claims 36 to 44 wherein the second component comprises: a composition comprising sildenafil, which composition is coated with a porous or semipermeable membrane suitable for promoting prolonged release of the sildenafil.
53. A pharmaceutical composition according to claim 52 wherein the semi-permeable membrane comprises a polymer.
54. A pharmaceutical composition according to claim 52 or claim 53 wherein the semipermeable membrane comprises a methacrylate polymer, ethylcellulose, cellulose acetate, poly(ethylene glycol), or a mixture of two or more thereof.
55. A pharmaceutical composition according to claim 54 wherein the semi-permeable membrane comprises cellulose acetate and poly(ethylene glycol).
56. A pharmaceutical composition according to any one of claims 52 to 55 wherein the composition comprising sildenafil which is coated with the membrane further comprises an osmagent.
57. A pharmaceutical composition according to claim 56 wherein the osmagent is selected from sodium chloride, potassium chloride, lithium chloride, magnesium chloride, mag- nesium sulphate, lithium sulphate, sodium sulphate, potassium sulphate, citric acid, mannitol, ribose, arabinose, galactose, leucine, glycine, fructose, sucrose, and sodium and other bicar- bonates, or a mixture of two or more thereof.
58. A pharmaceutical composition according to any one of claims 36 to 44 wherein the second component comprises: said sildenafil and a coating for delaying exposure of the sildenafil to buccal, gastric, or intestinal fluids.
59. A pharmaceutical composition according to claim 58 which comprises beadlets, pellets, spheroids, minitablets and/or granules comprising the sildenafil which are coated with said coating.
60. A pharmaceutical composition according to claim 58 or claim 59 wherein the coating comprises an enteric coating which dissolves at a pH greater than 5.0.
61. A pharmaceutical composition according to claim 60 wherein the enteric coating comprises a methacrylic acid-methyl methacrylate co-polymer.
62. A pharmaceutical composition according to claim 58 or claim 59 wherein the coating comprises a pH-dependent polymer.
63. A pharmaceutical composition according to claim 62 wherein the pH-dependent polymer is selected from cellulose acetate phthalate, hydroxypropylmethylcellulose phthalate 50, hydroxypropylmethylcellulose phthalate 55, polyvinylacetate phthalate, methacrylic acid-methyl methacrylate copolymer (1 : 1), methacrylic acid-methyl methacrylate copolymer (2: 1), methacrylic acid-ethyl acrylate copolymer (2: 1), hydroxypropylmethylcellulose acetate succinate, poly(methylvinylether/maleic acid) monoethylester, poly(methylvinylether/maleic acid)n-butyl ester, and shellac.
64. A pharmaceutical composition according to claim 58 or claim 59 wherein the coating is a non-pH-dependant degradable coating.
65. A pharmaceutical composition according to claim 64 wherein the coating comprises a non-pH-dependent polymer, optionally wherein the polymer is selected from: acacia, alginate, amylase, beeswax, carboxymethylcellulose, carnuba wax, cellulose acetate, cholesterol, ethyl- cellulose, fatty acids, gelatine, glyceryl behenate, glyceryl monostearate, glyceryl mono- distearate, glyceryl tripalmitate, hypromellose, hydroxypropylcellulose, hydrogenated vegeta- ble oil, lecithin, methylcellulose, paraffin wax, pectin, polyethylene glycol, polycaprolactone, polyglycolic acid, polylactic acid, polyglyclide-co-lactide co-polymers, polyvinylprroylidone, starch, stearic acid, stearyl alcohol, partially hydrogenated cottonseed oil/soyabean oil, partially hydrogenated palm oil, partially hydrogenated cottonseed oil, partially hydrogenated soyabean oil, partially hydrogenated castor oil, and polyethylene glycol 3350.
66. A pharmaceutical composition according to any one of the preceding claims, wherein the first component is adapted to deliver sildenafil rapidly to promote fast onset of action wherein the sildenafil is adapted in a way to increase its solubility, optionally wherein the sildenafil comprises a co-crystal of sildenafil, optionally wherein the co-crystal of sildenafil is a co-crystal as defined in any one of claims 95 to 103.
67. A pharmaceutical composition according to any one of the preceding claims, wherein a second component is adapted to deliver the sildenafil from dose to dose by providing the sildenafil in a modified release form.
68. A pharmaceutical composition according to any one of the preceding claims, wherein a second component is adapted to deliver the sildenafil from dose to dose by providing the sildenafil in a modified release form by admixing it with a polymer.
69. A pharmaceutical composition according to claim 2 wherein the third component is adapted to deliver the sildenafil from dose to dose within the therapeutic window.
70. A pharmaceutical composition according to claim 69, wherein in the third component, the sildenafil is adapted to release rapidly and over a long period of time so that the sildenafil is released from dose to dose within the therapeutic window.
71. A pharmaceutical composition according to any one of claims 2, 69 and 70 wherein:
the first component is as defined in any one of claims 7 to 35 and 66; and the second component is as defined in any one of claims 36 to 65, 67 and 68.
72. A pharmaceutical composition according to any one of claims 2 and 69 to 71 wherein the third component comprises a pharmaceutically acceptable salt of sildenafil, sildenafil free base, a sildenafil co-crystal, or a mixture of two or more thereof.
73. A pharmaceutical composition according to any one of claims 2 and 69 to 72 wherein the third component comprises sildenafil citrate, a sildenafil fatty acid salt as defined in any one of claims 105 to 108, sildenafil free base, a sildenafil co-crystal which is optionally as defined in any one of claims 95 to 103, or a mixture of two or more thereof.
74. A pharmaceutical composition according to any one of claims 2 and 69 to 73 wherein the sildenafil in the third component is sildenafil citrate.
75. A pharmaceutical composition according to any one of claims 72 to 74 wherein the third component further comprises one or more excipients as defined in any one of claims 45 to 65.
76. A pharmaceutical composition according to any one of claims 4 to 6, wherein the pharmaceutical composition comprises two components: said first component and said second component, wherein
the first component comprises sildenafil admixed with one or more excipients that facilitate immediate release; and
the second component comprises sildenafil admixed with one or more excipients that facilitate a delayed release,
wherein the first component comprises from 10% to 30% by weight of the total amount of sildenafil in the first and second components and the second component comprises from 90% to 70% by weight of the total amount of sildenafil in the first and second compo- nents.
77. A pharmaceutical composition according to any one of claims 4 to 6, wherein the pharmaceutical composition comprises two components: said first component and said second component, wherein
the first component comprises from lOmg to 30mg sildenafil, optionally 20mg sildenafil, admixed with one or more excipients that facilitate immediate release; and
the second component comprises from 90mg to 70mg of sildenafil, optionally 80mg of sildenafil, admixed with one or more excipients that facilitate a delayed release.
78. A pharmaceutical composition according to any one of claims 4 to 6, wherein the pharmaceutical composition comprises two components: said first component and said second component, wherein:
(A) the first component comprises from 5mg to 15mg sildenafil, optionally 1 Omg sildenafil, admixed with one or more excipients that facilitate immediate release; and
the second component comprises from 45mg to 35mg sildenafil, optionally 40mg sildenafil, admixed with one or more excipients that facilitate a delayed release; or
(B) the first component comprises from 2.5mg to 7.5mg sildenafil, optionally 5mg sildenafil, admixed with one or more excipients that facilitate immediate release; and
the second component comprises from 17.5mg to 22.5mg of sildenafil, optionally 20mg sildenafil, admixed with one or more excipients that facilitate a delayed release.
79. A pharmaceutical composition according to any one of claims 76 to 78 wherein: the first component is as further defined in any one of claims 13 to 35; and the second component is as further defined in any one of claims 41 to 65.
80. A pharmaceutical composition according to any one of claims 76 to 78, wherein the first component comprises said sildenafil in the form of sildenafil citrate which is adapted to be released rapidly by being admixed with excipients which facilitate an immediate release which excipients are conventional excipients.
81. A pharmaceutical composition according to any one of claims 76 to 78 and 80, wherein the second component comprises said sildenafil citrate admixed with a polymer to facilitate a delayed release which polymer is a conventional polymer used in delayed release formulations.
82. A pharmaceutical composition according to any one of claims 76 to 78 wherein the sildenafil in the first component is a co-crystal of sildenafil, optionally wherein the co-crystal is as defined in any one of claims 95 to 103.
83. A pharmaceutical composition according to any one of claims 76 to 78 wherein the sildenafil in the first component is sildenafil free base.
84. A pharmaceutical composition according to any one of claims 76 to 78 wherein the sildenafil in the first component is a sildenafil fatty acid salt as defined in any one of claims 105 to 108.
85. A pharmaceutical composition according to any one of claims 76 to 84 which is a solid dosage form.
86. A pharmaceutical composition according to any one of claims 76 to 85 which is a swallow tablet.
87. A pharmaceutical composition according to any one of claims 4 to 86 wherein the first component is disposed on a surface of the second component.
88. A pharmaceutical composition according to any one of claims 4 to 87 which has a core-shell structure wherein the second component defines a core and the first component is disposed on the surface of the second component to form a shell which surrounds the core.
89. A multi-component dosage form comprising sildenafil, said dosage form being adapted in a first way to provide rapid release of sildenafil into the bloodstream, said dosage form being adapted in a second way to further provide a maintenance dose of sildenafil within the therapeutic window, and the dosage form being adapted in a third way to provide a modified or delayed release format of the sildenafil product which lasts from dose to dose.
90. A multi-component dosage form according to claim 89 which is a swallow tablet.
91. A multi-component dosage form according to claim 89 or claim 90 which comprises a first component and a second component as defined in any one of claims 4 to 88, and optionally comprises a third component as defined in any one of claims 2, 69, 70 and 72 to 75.
92. A multi-component dosage form according to any one of claims 89 to 91 which comprises:
sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co- crystal is optionally as defined in any one of claims 95 to 103;
a fatty acid salt of sildenafil as defined in any one of claims 105 to 108, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and
said delayed release component, comprising a bolus dose of sildenafil, optionally wherein the sildenafil in the bolus dose is in the form of sildenafil citrate.
93. A multi-component dosage form according to any one of claims 89 to 91 which com- prises:
from 2.5mg to 25mg sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co-crystal is optionally as defined in any one of claims 95 to 103;
from 2.5mg to 25mg sildenafil in the form of a fatty acid salt of sildenafil as defined in any one of claims 105 to 108, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and said delayed release component, comprising a bolus dose of sildenafil, optionally wherein the bolus dose of sildenafil is from 20mg to 95mg sildenafil, and optionally wherein the sildenafil in the bolus dose is in the form of sildenafil citrate.
94. A multi-component dosage form according to any one of claims 89 to 91 which comprises:
from 2.5 to 5mg sildenafil in the form of a co-crystal suitable for rapid release of sildenafil, which co-crystal is optionally as defined in any one of claims 95 to 103;
from 2.5 to 15mg sildenafil in the form of a fatty acid salt of sildenafil as defined in any one of claims 105 to 108, suitable for delivering sildenafil between onset of action from said rapid release and onset of a bolus amount of a delayed release component; and
said delayed release component, comprising a bolus dose of sildenafil, optionally wherein the sildenafil in the bolus dose is in the form of sildenafil citrate and optionally wherein the bolus dose is from lOmg to 80mg sildenafil, for instance 20mg sildenafil.
95. A co-crystal, which co-crystal comprises sildenafil and a co-crystal former, which co- crystal former is a compound which comprises a phenol moiety.
96. A co-crystal according to claim 95 wherein the co-crystal former is a compound of formula (I)

R is H, -C(0)OR2, -L-C(0)OR2, Ar or -L-Ar;
R2 is H or unsubstituted C alkyl;
L is -CH=CH-
Ar is phenyl or naphthyl, which phenyl or naphthyl is unsubstituted or substituted with from 1 to 3 groups selected from OH and C alkyl; or
Ar is a group of formula (II)

wherein
R3 is H or OH, and
m is 0, 1 or 2.
97. A co-crystal according to claim 96 wherein, when R is H, n is 2 or 3.
98. A co-crystal according to claim 96 or claim 97 wherein R2 is H or methyl; Ar is phenyl, which phenyl is unsubstituted or substituted with from 1 to 3 OH groups; or Ar is said group of formula (II) wherein R3 is OH.
99. A co-crystal according to any one of claims 95 to 98 wherein the co-crystal former is selected from compounds having the following structures:

100. A co-crystal according to any one of claims 95 to 98 wherein the co-crystal former is selected from compounds having the following structures:

wherein the molar ratio of the sildenafil to the co-crystal former in the co-crystal is 1 : 1.
101. A co-crystal according to any one of claims 95 to 98 wherein the co-crystal former llowing structure:

wherein the molar ratio of the sildenafil to the co-crystal former in the co-crystal is 3:2.
102. A co-crystal according to any one of claims 95 to 101 wherein the sildenafil in the co crystal is sildenafil base, i.e. 5- {2-ethoxy-5-[(4-methylpiperazin-l -yl)sulfonyl]phenyl}-l- me thyl-3-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one.
103. A co-crystal according to any one of claims 95 to 102 which is co-crystal P42-I-A,
P42-I-B, P42-III, P42-VI-B, P42-VI-C, P42-VI-D, P42-VI-E, P42-VII, P42-VIII P42-IX as described herein.
104. A pharmaceutical composition comprising: (i) a co-crystal as defined in any one of claims 95 to 103, and (ii) a pharmaceutically acceptable carrier.
105. A salt of sildenafil with a long chain fatty acid.
106. A salt according to claim 105 which is amorphous or crystalline.
107. A salt according to claim 105 or claim 106 wherein the long chain fatty acid has for- mula R-C(0)OH wherein R is Ce-24 alkyl or Ce-24 alkenyl.
108. A salt according to any one of claims 105 to 107 wherein the long chain fatty acid is selected from decanoic acid, docosanoic acid, eicosanoic acid, heneicosanoic acid, heptadeca- noic acid, lauric acid, myristic acid, nonadecanoic acid, nonanoic acid, octanoic acid, palmitic
111
acid, pentadecanoic acid, stearic acid, tetracosanoic acid, tricosanoic acid, tridecanoic acid, undecanoic acid, and undecylenic acid.
109. A pharmaceutical composition comprising: (i) a salt of sildenafil with a long chain fatty acid as defined in any one of claims 105 to 108, and (ii) a pharmaceutically acceptable carrier.
110. A pharmaceutical composition according to claim 109 which comprises (i) said salt of sildenafil and (ii) a polymeric carrier or matrix material.
111. A pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, for use in a method of treatment of the human or animal body by therapy.
112. A pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, for use in the treatment or prophylaxis of a disorder associated with PDE5 inhibition.
113. Use of a pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co- crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, in the manufacture of a medicament for use in the treatment or prophylaxis of a disor- der associated with PDE5 inhibition.
114. A method for the treatment or prophylaxis of a disorder associated with PDE5 inhibition, which method comprises administering an effective amount of a pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, to a sufferer in need thereof.
115. A pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, for use in the treatment of human male erectile dysfunction.
116. Use of a pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co- crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, in the manufacture of a medicament for use in the treatment of human male erectile dysfunction.
117. A method for the treatment of human male erectile dysfunction, which method comprises administering an effective amount of a pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, to a human male in need thereof.
118. A pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, for use in the treatment of human female sexual dysfunction.
119. Use of a pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co- crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, in the manufacture of a medicament for use in the treatment of human female sexual dysfunction.
120. A method for the treatment of human female sexual dysfunction, which method com- prises administering an effective amount of a pharmaceutical composition as defined in any one of claims 1 to 88, 104, 109 and 110, a multi-component dosage form as defined in any one of claims 89 to 94, a co-crystal as defined in any one of claims 95 to 103, or a salt as defined in any one of claims 105 to 108, to a human female in need thereof.
121. A pharmaceutical composition, a multi-component dosage form, co-crystal or salt according to claim 115 or claim 118 for use as defined in said claim, the use according to claim 116 or claim 119, or a method according to claim 117 or claim 120, wherein:
said treatment comprises delivering sildenafil rapidly to promote fast onset of action, and delivering sildenafil from dose to dose within the therapeutic window.
122. A pharmaceutical composition, a multi-component dosage form, co-crystal or salt according to claim 115, 118 or 121 for use as defined in said claim, the use according to claim 116, 119 or 121, or a method according to claim 117, 120 or 121, wherein:
said treatment comprises providing rapid release of sildenafil into the bloodstream, providing a maintenance dose of sildenafil within the therapeutic window, and providing modified or delayed release of sildenafil to last from dose to dose.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1605714.3 | 2016-04-02 | ||
| GB201605714 | 2016-04-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017168174A1 true WO2017168174A1 (en) | 2017-10-05 |
Family
ID=58503660
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2017/050921 WO2017168174A1 (en) | 2016-04-02 | 2017-03-31 | New pharmaceutical forms of sildenafil |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017168174A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019081451A1 (en) * | 2017-10-26 | 2019-05-02 | Mw Encap Limited | Liquid filled formulations of pde5 inhibitors |
| CN111388433A (en) * | 2020-03-27 | 2020-07-10 | 广州白云山医药集团股份有限公司白云山制药总厂 | Sildenafil citrate oral preparation and preparation method thereof |
| JP2021054781A (en) * | 2019-09-26 | 2021-04-08 | デボン エルエス,リミテッド | Co-crystalline efinaconazole and method for producing the same |
| CN113694035A (en) * | 2021-10-09 | 2021-11-26 | 药大制药有限公司 | Preparation method of sildenafil tablets |
Citations (251)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3760984A (en) | 1971-09-29 | 1973-09-25 | Alza Corp | Osmotically powered agent dispensing device with filling means |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US3987790A (en) | 1975-10-01 | 1976-10-26 | Alza Corporation | Osmotically driven fluid dispenser |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US4036227A (en) | 1973-04-25 | 1977-07-19 | Alza Corporation | Osmotic releasing device having a plurality of release rate patterns |
| US4576604A (en) | 1983-03-04 | 1986-03-18 | Alza Corporation | Osmotic system with instant drug availability |
| US4578075A (en) | 1982-12-20 | 1986-03-25 | Alza Corporation | Delivery system housing a plurality of delivery devices |
| US4642903A (en) | 1985-03-26 | 1987-02-17 | R. P. Scherer Corporation | Freeze-dried foam dosage form |
| US4673405A (en) | 1983-03-04 | 1987-06-16 | Alza Corporation | Osmotic system with instant drug availability |
| US4681583A (en) | 1982-12-20 | 1987-07-21 | Alza Corporation | System for dispersing drug in biological environment |
| US4693895A (en) | 1984-10-26 | 1987-09-15 | Alza Corporation | Colon delivery system |
| US4705515A (en) | 1984-10-26 | 1987-11-10 | Alza Corporation | Dosage form for administering drug of the colon |
| US4773907A (en) | 1982-12-20 | 1988-09-27 | Alza Corporation | Primary delivery system comprising secondary dosage form |
| US4855326A (en) | 1987-04-20 | 1989-08-08 | Fuisz Pharmaceutical Ltd. | Rapidly dissoluble medicinal dosage unit and method of manufacture |
| EP0463756A1 (en) | 1990-06-20 | 1992-01-02 | Pfizer Limited | Pyrazolopyrimidinone antianginal agents |
| US5178878A (en) | 1989-10-02 | 1993-01-12 | Cima Labs, Inc. | Effervescent dosage form with microparticles |
| US5229133A (en) | 1990-01-24 | 1993-07-20 | Alza Corporation | Delivery system comprising means for controlling internal pressure |
| US5250534A (en) | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5464632A (en) | 1991-07-22 | 1995-11-07 | Laboratoires Prographarm | Rapidly disintegratable multiparticular tablet |
| US5576014A (en) | 1994-01-31 | 1996-11-19 | Yamanouchi Pharmaceutical Co., Ltd | Intrabuccally dissolving compressed moldings and production process thereof |
| US5607697A (en) | 1995-06-07 | 1997-03-04 | Cima Labs, Incorporated | Taste masking microparticles for oral dosage forms |
| US5738875A (en) | 1994-10-28 | 1998-04-14 | R.P. Scherer Corporation | Process for preparing solid pharmaceutical dosage forms |
| US5858694A (en) | 1997-05-30 | 1999-01-12 | Cell Pathways, Inc. | Method for identifying compounds for inhibition of cancerous lesions |
| US5874437A (en) | 1996-11-01 | 1999-02-23 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US5955611A (en) | 1996-06-14 | 1999-09-21 | Pfizer Inc. | Process for preparing sildenafil |
| US5958926A (en) | 1996-11-01 | 1999-09-28 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US5981563A (en) | 1995-04-28 | 1999-11-09 | Zonagen, Inc. | Methods and formulations for modulating the human sexual response |
| US6007824A (en) | 1998-07-09 | 1999-12-28 | Duckett; Melvin J. | Natural composition and method for the treatment of sexual dysfunction |
| US6013663A (en) | 1997-04-02 | 2000-01-11 | Sankyo Company, Limited | Dithiolan derivatives, their preparation and their therapeutic effect |
| US6037346A (en) | 1997-10-28 | 2000-03-14 | Vivus, Inc. | Local administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6043252A (en) | 1997-05-05 | 2000-03-28 | Icos Corporation | Carboline derivatives |
| WO2000024383A1 (en) * | 1998-10-23 | 2000-05-04 | Pfizer Research And Development Company, N.V./S.A. | Controlled-release pharmaceutical formulations containing a cgmp pde-5 inhibitor |
| US6075028A (en) | 1999-09-23 | 2000-06-13 | Graham; Richard | Method of treating Tourette's syndrome and related CNS disorders |
| US6077841A (en) | 1998-09-04 | 2000-06-20 | Janssen Pharmaceutica, N.V. | 5-heterocyclyl pyrazolo[4,3-d]pyrimidin-7-ones for the treatment of male erectile dysfunction |
| US6087368A (en) | 1998-06-08 | 2000-07-11 | Bristol-Myers Squibb Company | Quinazolinone inhibitors of cGMP phosphodiesterase |
| US6087362A (en) | 1999-03-16 | 2000-07-11 | Pentech Pharmaceuticals, Inc. | Apomorphine and sildenafil composition |
| US6102849A (en) | 1999-04-03 | 2000-08-15 | Hakac; John R. | Non-surgical penile prosthesis |
| US6124461A (en) | 1998-05-26 | 2000-09-26 | Saint Louis University, Health Services Center, Research Administration | Compounds, compositions, and methods for treating erectile dysfunction |
| US6127363A (en) | 1997-10-28 | 2000-10-03 | Vivus, Inc. | Local administration of Type IV phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6130053A (en) | 1999-08-03 | 2000-10-10 | Cell Pathways, Inc. | Method for selecting compounds for inhibition of neoplastic lesions |
| US6132757A (en) | 1998-05-01 | 2000-10-17 | Neal R. Cutler | Treatment of sexual dysfunction in certain patient groups |
| US6143757A (en) | 1995-07-14 | 2000-11-07 | Icos Corporation | Chemical compounds |
| US6143746A (en) | 1994-01-21 | 2000-11-07 | Icos Corporation | Tetracyclic cyclic GMP-specific phosphodiesterase inhibitors, process of preparation and use |
| US6156753A (en) | 1997-10-28 | 2000-12-05 | Vivus, Inc. | Local administration of type III phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6165975A (en) | 1997-06-23 | 2000-12-26 | Queen's University At Kingston | Microdose therapy |
| US6184231B1 (en) | 1998-12-04 | 2001-02-06 | Bristol-Myers Squibb | 3-substituted-4-arylquinolin-2-one derivatives as potassium channel modulators |
| US6187790B1 (en) | 1999-03-04 | 2001-02-13 | Neal R. Cutler | Use of cilostazol for treatment of sexual dysfunction |
| US6194433B1 (en) | 1998-10-05 | 2001-02-27 | Neal R. Cutler | Sexual dysfunction in females |
| US6200771B1 (en) | 1998-10-15 | 2001-03-13 | Cell Pathways, Inc. | Method of using a novel phosphodiesterase in pharmaceutical screeing to identify compounds for treatment of neoplasia |
| US6200591B1 (en) | 1998-06-25 | 2001-03-13 | Anwar A. Hussain | Method of administration of sildenafil to produce instantaneous response for the treatment of erectile dysfunction |
| US6200571B1 (en) | 1996-06-10 | 2001-03-13 | Lotfi Ismail Omar | Methods and compositions for the treatment of sexual dysfunction in humans and animals |
| US6204383B1 (en) | 1998-05-15 | 2001-03-20 | Torcan Chemical Ltd. | Processes for preparing sildenafil |
| US6207829B1 (en) | 1998-10-12 | 2001-03-27 | Pfizer Inc. | Process for preparation of pyrazolo[4,3-d]pyrimidin-7-ones and intermediates thereof |
| US6211156B1 (en) | 1999-11-10 | 2001-04-03 | Asta Medica A.G. | Peptides for treatment of erectile dysfunction |
| US6214849B1 (en) | 1999-04-29 | 2001-04-10 | Lupin Laboratories Limited | Use of nicorandil in treatment of sexual dysfunction or for enhancement of sexual function in mammals including humans |
| US6221402B1 (en) | 1997-11-20 | 2001-04-24 | Pfizer Inc. | Rapidly releasing and taste-masking pharmaceutical dosage form |
| US6235776B1 (en) | 1998-11-12 | 2001-05-22 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a paclitaxel derivative |
| US6235782B1 (en) | 1998-11-12 | 2001-05-22 | Rifat Pamukcu | Method for treating a patient with neoplasia by treatment with a platinum coordination complex |
| US6239117B1 (en) | 1997-02-13 | 2001-05-29 | Albert Einstein College Of Medicine Of Yeshiva University | Gene therapy for regulating bladder smooth muscle tone |
| US6241752B1 (en) | 1998-05-08 | 2001-06-05 | Inventis | Method of treating for impotence and apparatus particularly useful in such method |
| US6242444B1 (en) | 1999-06-04 | 2001-06-05 | The Jordanian Pharmaceutical Manufacturing And Medical Equipment Co., Ltd. | Compounds and pharmaceutical compositions containing the same |
| USRE37234E1 (en) | 1996-11-01 | 2001-06-19 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiestrase inhibitor compounds, compositions and their uses |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6251428B1 (en) | 1998-07-24 | 2001-06-26 | Seo Hong Yoo | Preparation of aqueous clear solution dosage forms with bile acids |
| US6251436B1 (en) | 1995-09-29 | 2001-06-26 | L.A.M. Pharmaceutical Corporation | Drug preparations for treating sexual dysfunction |
| US6266560B1 (en) | 1998-06-19 | 2001-07-24 | Genetronics, Inc. | Electrically assisted transdermal method and apparatus for the treatment of erectile dysfunction |
| US6265420B1 (en) | 1998-06-23 | 2001-07-24 | Medinox, Inc. | Use of nitric oxide scavengers to treat side effects caused by therapeutic administration of sources of nitric oxide |
| US6268338B1 (en) | 1993-04-30 | 2001-07-31 | Merck & Co., Inc. | Cyclohexapeptidyl amine compounds |
| US6267985B1 (en) | 1999-06-30 | 2001-07-31 | Lipocine Inc. | Clear oil-containing pharmaceutical compositions |
| US6271228B1 (en) | 2000-04-28 | 2001-08-07 | Pfizer Inc. | Blood pressure stabilization during hemodialysis |
| US6271211B1 (en) | 1997-02-13 | 2001-08-07 | Albert Einstein College Of Medicine Of Yeshiva University | Gene therapy for regulating penile smooth muscle tone |
| US6277884B1 (en) | 1998-06-01 | 2001-08-21 | Nitromed, Inc. | Treatment of sexual dysfunction with N-hydroxyguanidine compounds |
| US6284763B1 (en) | 1998-08-26 | 2001-09-04 | Queen's University At Kingston | Methods for remodeling neuronal and cardiovascular pathways |
| US6291528B1 (en) | 1991-07-03 | 2001-09-18 | Nathan Earl Scott | Prostaglandin E1/F2 in combination with prostaglandin F2α for enhancing female sexual arousal |
| US6290986B1 (en) | 1996-10-24 | 2001-09-18 | Pharmaceutical Applications Associates, Llc | Method and composition for transdermal administration of pharmacologic agents |
| US6291471B1 (en) | 1998-12-17 | 2001-09-18 | Abb Holdings, Inc. | Use of apomorphine for the treatment of organic erectile dysfunction in males |
| US6294192B1 (en) | 1999-02-26 | 2001-09-25 | Lipocine, Inc. | Triglyceride-free compositions and methods for improved delivery of hydrophobic therapeutic agents |
| US6294550B1 (en) | 1997-10-28 | 2001-09-25 | Asivi, Llc | Treatment of female sexual dysfunction |
| US6294534B1 (en) | 1998-06-11 | 2001-09-25 | Merck & Co., Inc. | Spiropiperidine derivatives as melanocortin receptor agonists |
| US6303606B1 (en) | 1999-05-07 | 2001-10-16 | Recordati, S.A., Chemical And Pharmaceutical Company | Use of selective antagonists of the α1B-adrenergic receptor for improvement of sexual dysfunction |
| US6303135B1 (en) | 1999-07-08 | 2001-10-16 | Neal R. Cutler | Use of quinolines and quinolones to treat male erectile dysfunction |
| US6316438B1 (en) | 1999-03-22 | 2001-11-13 | Bristol-Myers Squibb Co. | Fused pyridopyridazine inhibitors of cGMP phosphodiesterase |
| US6323242B1 (en) | 1998-12-02 | 2001-11-27 | Peter Sterling Mueller | Treatment of disorders secondary to organic impairments |
| US6326379B1 (en) | 1998-09-16 | 2001-12-04 | Bristol-Myers Squibb Co. | Fused pyridine inhibitors of cGMP phosphodiesterase |
| US6331543B1 (en) | 1996-11-01 | 2001-12-18 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitors, compositions and methods of use |
| US6333354B1 (en) | 1997-02-28 | 2001-12-25 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Synergistic combination of PDE inhibitors and adenylate cyclase agonists or guanyl cyclyse agonists |
| US6338862B1 (en) | 2001-03-26 | 2002-01-15 | Sarfaraz K Niazi | Composition and method of use in treating sexual dysfunction using cGMP-specific phosphodiesterase type 5 inhibitors |
| US6342251B1 (en) | 1997-12-02 | 2002-01-29 | West Pharmaceutical Services Drug Delivery & Clinical Research Centre Limited | Compositions for nasal administration |
| US6346271B1 (en) | 1995-09-29 | 2002-02-12 | L. A. M. Pharmaceutical Corporation | Topical drug preparations |
| US6350760B1 (en) | 1999-06-04 | 2002-02-26 | Merck & Co., Inc. | Substituted piperidines as melanocortin-4 receptor agonists |
| US6362178B1 (en) | 1997-11-12 | 2002-03-26 | Bayer Aktiengesellschaft | 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| US6365590B1 (en) | 1998-05-26 | 2002-04-02 | Saint Louis University | Compounds, compositions and methods for treating erectile dysfunction |
| US6368640B1 (en) | 1998-04-03 | 2002-04-09 | The Daily Wellness Company | Method and composition for improving sexual fitness |
| US6376554B1 (en) | 1999-03-19 | 2002-04-23 | Knoll Pharmaceutical Company | Method of treating sexual dysfunction |
| US6376509B2 (en) | 2000-05-30 | 2002-04-23 | Merck & Co., Inc. | Melanocortin receptor agonists |
| US6380267B1 (en) | 1999-09-13 | 2002-04-30 | David M. Swope | Composition and method for decreasing neurologic symptomatology |
| US6383789B1 (en) | 2001-03-22 | 2002-05-07 | Pe Corporation (Ny) | Isolated human UDP-glycosyltransferase, nucleic acid molecules encoding human UDP-glycosyltransferase, and uses thereof |
| US6383471B1 (en) | 1999-04-06 | 2002-05-07 | Lipocine, Inc. | Compositions and methods for improved delivery of ionizable hydrophobic therapeutic agents |
| US6387407B1 (en) | 1995-09-29 | 2002-05-14 | L.A.M. Pharmaceutical Corporation | Topical drug preparations |
| US6391869B1 (en) | 1998-12-14 | 2002-05-21 | Cellergy Pharmaceuticals, Inc. | Compositions and methods for the treatment of anorectal disorders |
| US6395300B1 (en) | 1999-05-27 | 2002-05-28 | Acusphere, Inc. | Porous drug matrices and methods of manufacture thereof |
| US6395736B1 (en) | 1998-12-14 | 2002-05-28 | Cellegy Pharmaceuticals, Inc. | Compositions and methods for the treatment of anorectal disorders |
| US6395744B1 (en) | 1994-04-22 | 2002-05-28 | Queen's University At Kingston | Method and compositions for the treatment or amelioration of female sexual dysfunction |
| US6399579B1 (en) | 2000-08-15 | 2002-06-04 | Hauser, Inc. | Compositions comprising icariside I and anhydroicaritin and methods for making the same |
| US6399601B1 (en) | 1999-09-30 | 2002-06-04 | Pfizer Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
| US6403597B1 (en) | 1997-10-28 | 2002-06-11 | Vivus, Inc. | Administration of phosphodiesterase inhibitors for the treatment of premature ejaculation |
| US6403658B1 (en) | 2000-09-21 | 2002-06-11 | Shaina Toppo | Genital vasodilator |
| US6407259B1 (en) | 2000-07-28 | 2002-06-18 | Pfizer Inc. | Process for the preparation of pyrazoles |
| US6410595B1 (en) | 1997-07-09 | 2002-06-25 | Androsolutions, Inc. | Methods and compositions for treating male erectile dysfunction |
| US6414027B1 (en) | 1997-07-09 | 2002-07-02 | Androsolutions, Inc. | Methods and compositions for treating male erectile dysfunction |
| US6413496B1 (en) | 1996-12-04 | 2002-07-02 | Biogland Ireland (R&D) Limited | Pharmaceutical compositions and devices for their administration |
| US6413968B1 (en) | 2000-06-30 | 2002-07-02 | O'donnell, Jr. Francis E. | Treatment of myasthenia gravis by cyclic nucleotide phosphodiesterase inhibition |
| US6417207B1 (en) | 1999-05-12 | 2002-07-09 | Nitromed, Inc. | Nitrosated and nitrosylated potassium channel activators, compositions and methods of use |
| US6417208B1 (en) | 1999-02-05 | 2002-07-09 | Albert Einstein College Of Medicine Of Yeshiva University | Method of identification of inhibitors of PDE1C |
| US6420150B1 (en) | 2000-11-27 | 2002-07-16 | Pe Corporation (Ny) | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6426084B1 (en) | 2000-06-19 | 2002-07-30 | Neal R. Cutler | Treatment of sexual dysfunction in certain patient groups |
| US6428769B1 (en) | 1999-05-04 | 2002-08-06 | Aradigm Corporation | Acute testosterone administration |
| US6436944B1 (en) | 1999-09-30 | 2002-08-20 | Pfizer Inc. | Combination effective for the treatment of impotence |
| US6436997B1 (en) | 1998-06-01 | 2002-08-20 | Nitromed, Inc. | Endogenous nitric oxide synthesis under conditions of low oxygen tension |
| US6436684B1 (en) | 2000-03-27 | 2002-08-20 | Applera Corporation | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6444237B1 (en) | 2001-09-13 | 2002-09-03 | Pamela A. Heleen | Herbal composition for enhancing sexual response |
| US6443152B1 (en) | 2001-01-12 | 2002-09-03 | Becton Dickinson And Company | Medicament respiratory delivery device |
| US6448293B1 (en) | 2000-03-31 | 2002-09-10 | Pfizer Inc. | Diphenyl ether compounds useful in therapy |
| US6451807B1 (en) | 1999-04-30 | 2002-09-17 | Lilly Icos, Llc. | Methods of treating sexual dysfunction in an individual suffering from a retinal disease, class 1 congestive heart failure, or myocardial infarction using a PDE5 inhibitor |
| US6451813B1 (en) | 2001-01-26 | 2002-09-17 | R. T. Alamo Ventures I, Llc | Treatment of gastroparesis in certain patient groups |
| US6455654B1 (en) | 2000-09-01 | 2002-09-24 | Samsung Electronics Co., Ltd. | Water-soluble polymeric adhesion promoter and production method |
| US6455702B1 (en) | 2001-05-16 | 2002-09-24 | Aims Fine Chemicals, Inc. | Process for the production of N,N-carbonyl diimidazole |
| US6455572B1 (en) | 2000-04-07 | 2002-09-24 | Pfizer Inc. | Estrogen agonist/antagonist metabolites |
| US6458804B1 (en) | 2001-01-26 | 2002-10-01 | R.T. Alamo Venturesi, Llc | Methods for the treatment of central nervous system disorders in certain patient groups |
| US6458790B2 (en) | 2000-03-23 | 2002-10-01 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| US6462047B1 (en) | 1998-09-16 | 2002-10-08 | Icos Corporation | Carboline derivatives as cGMP phosphodiesterase inhibitors |
| US6465494B1 (en) | 2001-08-24 | 2002-10-15 | Cell Pathways, Inc. | Methods for treatment of cystic fibrosis |
| US6465465B1 (en) | 2000-03-14 | 2002-10-15 | Arzneimittelwerk Dresden Gmbh | Process for the treatment of erectile dysfunction and product therefor |
| US6469024B2 (en) | 2000-05-11 | 2002-10-22 | Bristol-Myers Squibb Company | Tetrahydroisoquinoline analogs useful as growth hormone secretagogues |
| US6469012B1 (en) | 1993-06-09 | 2002-10-22 | Pfizer Inc | Pyrazolopyrimidinones for the treatment of impotence |
| US6469065B1 (en) | 1996-02-02 | 2002-10-22 | Nitromed, Inc. | Nitrosated and nitrosylated α-adrenergic receptor antagonist, compositions and methods of use |
| US6472425B1 (en) | 1997-10-31 | 2002-10-29 | Nitromed, Inc. | Methods for treating female sexual dysfunctions |
| US6472420B2 (en) | 1998-11-12 | 2002-10-29 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a paclitaxel derivative |
| US6476037B1 (en) | 2000-03-23 | 2002-11-05 | The Regents Of The University Of California | L-arginine and phosphodiesterase (PDE) inhibitor synergism |
| US6476074B1 (en) | 1997-07-10 | 2002-11-05 | Synphora Ab | Method and composition for treatment of erectile dysfunction |
| US6476021B1 (en) | 1997-11-28 | 2002-11-05 | Mochida Pharmaceutical Co., Ltd. | Compounds having cGMP-PDE inhibitory effect |
| US6477410B1 (en) | 2000-05-31 | 2002-11-05 | Biophoretic Therapeutic Systems, Llc | Electrokinetic delivery of medicaments |
| US6479074B2 (en) | 1996-10-24 | 2002-11-12 | Pharmaceutical Applications Associates Llc | Methods and transdermal compositions for pain relief |
| US6479493B1 (en) | 2001-08-23 | 2002-11-12 | Cell Pathways, Inc. | Methods for treatment of type I diabetes |
| US6482426B1 (en) | 1998-09-17 | 2002-11-19 | Zonagen, Inc. | Compositions for the treatment of male erectile dysfunction |
| US6482948B1 (en) | 1999-03-30 | 2002-11-19 | Nippon Soda Co., Ltd. | Thienopyrimidine compounds and salts thereof and process for the preparation of the same |
| US6482398B1 (en) | 1995-06-07 | 2002-11-19 | The Procter & Gamble Company | Transfer-resistant lip compositions |
| US6485747B1 (en) | 1998-10-30 | 2002-11-26 | Monsanto Company | Coated active tablet(s) |
| US6492358B2 (en) | 2000-05-17 | 2002-12-10 | Ortho-Mcneil Pharmaceutical, Inc. | β-carboline derivatives useful as inhibitors of phosphodiesterase |
| US6492371B2 (en) | 2000-04-19 | 2002-12-10 | H H. Roylance | Use of cyclic GMP-specific phosphodiesterase inhibitors for treatment of Parkinson's Disease |
| US6497885B2 (en) | 2000-12-22 | 2002-12-24 | The Daily Wellness Company | Method and composition for improving fertility health in female and male animals and humans |
| WO2002102802A1 (en) | 2001-06-14 | 2002-12-27 | Korea Institute Of Science And Technology | Novel pyrazolopyrimidinethione derivatives. preparation methods thereof and their use as therapeutics for erectile dysfunction |
| US6500610B1 (en) | 1997-05-30 | 2002-12-31 | Cell Pathways, Inc | Methods for identifying compounds for inhibiting of neoplastic lesions, and pharmaceutical compositions containing such compounds |
| US6500440B1 (en) | 2000-06-23 | 2002-12-31 | Whan In Pharm Co., Ltd. | Topical preparation of alprostadil for the treatment of erectile dysfunction |
| US6499984B1 (en) | 2000-05-22 | 2002-12-31 | Warner-Lambert Company | Continuous production of pharmaceutical granulation |
| US6512002B2 (en) | 2000-01-12 | 2003-01-28 | Pfizer Inc. | Methods of treatment for premature ejaculation in a male |
| US6511973B2 (en) | 2000-08-02 | 2003-01-28 | Bristol-Myers Squibb Co. | Lactam inhibitors of FXa and method |
| US6528521B2 (en) | 2000-11-15 | 2003-03-04 | Tap Pharmaceutical Products, Inc. | Treatment of anti-depression drug-induced sexual dysfunction with apomorphine |
| US20030044457A1 (en) * | 2001-07-17 | 2003-03-06 | Joaquina Faour | Drug delivery device containing oseltamivir and an H1 antagonist |
| US6531114B1 (en) | 1999-04-06 | 2003-03-11 | Wm. Wrigley Jr. Company | Sildenafil citrate chewing gum formulations and methods of using the same |
| US6531297B2 (en) | 2000-10-20 | 2003-03-11 | Applera Corporation | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6541638B2 (en) | 2000-07-19 | 2003-04-01 | Hoffman-La Roche Inc. | Pyrrolidine derivatives |
| US6541487B1 (en) | 1998-05-01 | 2003-04-01 | R.T. Alamo Ventures I, Llc | PDE III inhibitors for treating sexual dysfunction |
| US6544981B2 (en) | 2000-06-09 | 2003-04-08 | Bristol-Myers Squibb Company | Lactam inhibitors of factor Xa and method |
| US6548087B1 (en) | 1999-07-12 | 2003-04-15 | Frances B. Kent | Nutritional supplement |
| US6548490B1 (en) | 1997-10-28 | 2003-04-15 | Vivus, Inc. | Transmucosal administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6548044B1 (en) | 1999-11-22 | 2003-04-15 | Epix Medical, Inc. | Imaging sexual response |
| US6548508B2 (en) | 2000-10-20 | 2003-04-15 | Pfizer, Inc. | Use of PDE V inhibitors for improved fecundity in mammals |
| US6548544B1 (en) | 1999-11-18 | 2003-04-15 | The National University Of Singapore | Method of treatment of priapism without raising systemic blood pressure |
| US6552024B1 (en) | 1999-01-21 | 2003-04-22 | Lavipharm Laboratories Inc. | Compositions and methods for mucosal delivery |
| US6555547B1 (en) | 2000-02-28 | 2003-04-29 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a vinca alkaloid derivative |
| US6562838B2 (en) | 2001-01-26 | 2003-05-13 | R. T. Alamo Ventures I, L.L.C. | Treatment of cardiovascular disease with quinolinone enantiomers |
| US6562868B1 (en) | 1999-01-08 | 2003-05-13 | Synphora Ab | Method for treatment of female sexual dysfunction |
| US6565851B2 (en) | 2000-05-26 | 2003-05-20 | Horphag Research Limited | Relieving symptoms of erectile dysfunction with proanthocyanidins |
| US6569638B1 (en) | 2000-03-03 | 2003-05-27 | Cell Pathways, Inc | Method for screening compounds for the treatment of neoplasia |
| US6569123B2 (en) | 1999-10-14 | 2003-05-27 | Becton, Dickinson And Company | Prefillable intradermal injector |
| US6569143B2 (en) | 1999-10-14 | 2003-05-27 | Becton, Dickinson And Company | Method of intradermally injecting substances |
| US6572880B2 (en) | 1996-10-24 | 2003-06-03 | Pharmaceutical Applications Associates Llc | Methods and transdermal compositions for pain relief |
| US6573285B2 (en) | 2000-12-21 | 2003-06-03 | Bristol-Myers Squibb Co. | Method for preventing or treating pain by administering an endothelin antagonist |
| US6576644B2 (en) | 2000-09-06 | 2003-06-10 | Bristol-Myers Squibb Co. | Quinoline inhibitors of cGMP phosphodiesterase |
| US6579968B1 (en) | 1999-06-29 | 2003-06-17 | Palatin Technologies, Inc. | Compositions and methods for treatment of sexual dysfunction |
| US6579879B2 (en) | 2001-03-30 | 2003-06-17 | Pfizer Inc | Pyridazinone aldose reductase inhibitors |
| US6583147B1 (en) | 1998-11-11 | 2003-06-24 | Dong A Pharm Co., Ltd. | Pyrazolopyrimidinone derivatives for the treatment of impotence |
| US6585958B1 (en) | 1998-07-24 | 2003-07-01 | Jago Research Ag | Medicinal aerosol formulations |
| US6586478B2 (en) | 2000-02-22 | 2003-07-01 | Cellegy Canada | Methods and compositions for improving sleep |
| US6589990B1 (en) | 1997-05-06 | 2003-07-08 | Panagiotis Kanakaris | Methods and compositions for misoprostol compound treatment of erectile dysfunction |
| US6593369B2 (en) | 1997-10-20 | 2003-07-15 | Vivus, Inc. | Methods, compositions, and kits for enhancing female sexual desire and responsiveness |
| US6596900B2 (en) | 2001-04-19 | 2003-07-22 | Pfizer Inc | Fused bicyclic or tricyclic amino acids |
| US6605627B2 (en) | 1995-06-07 | 2003-08-12 | The Salk Insitute For Biological Studies | Modulators of peroxisome proliferator activated receptor-gamma, and methods for the use thereof |
| US6604698B2 (en) | 2000-05-10 | 2003-08-12 | Skyepharma Canada, Inc. | Media milling |
| US6610747B2 (en) | 2000-08-31 | 2003-08-26 | Pfizer Inc. | Phenoxybenzylamine derivatives as SSRIs |
| US6613768B1 (en) | 1999-04-30 | 2003-09-02 | Lilly Icos Llc | Treatment of female arousal disorder |
| US6613344B2 (en) | 2000-09-28 | 2003-09-02 | Nanocyte Inc. | Methods utilizing stinging cells/capsules |
| US6623768B1 (en) | 2002-04-16 | 2003-09-23 | Yousry M. A. Naguib | Pharmaceutically active composition extracted from Ferula hermonis and process of its extraction |
| US6622721B2 (en) | 1999-10-14 | 2003-09-23 | Becton, Dickinson And Company | Drug delivery system including holder and drug container |
| US6624138B1 (en) | 2001-09-27 | 2003-09-23 | Gp Medical | Drug-loaded biological material chemically treated with genipin |
| US6627632B2 (en) | 1998-12-14 | 2003-09-30 | Cellegy Pharmaceuticals, Inc. | Compositions and methods for the treatment of anorectal disorders |
| US6627234B1 (en) | 1998-12-15 | 2003-09-30 | Wm. Wrigley Jr. Company | Method of producing active agent coated chewing gum products |
| US6630504B2 (en) | 2000-08-31 | 2003-10-07 | Pfizer Inc. | Phenoxyphenylheterocyclyl derivatives as SSRIs |
| US6632419B2 (en) | 1999-05-04 | 2003-10-14 | Aradigm Corporation | Increasing libido in humans via acute testosterone administration |
| US6635274B1 (en) | 2000-10-27 | 2003-10-21 | Biochemics, Inc. | Solution-based transdermal drug delivery system |
| US6634576B2 (en) | 2000-08-31 | 2003-10-21 | Rtp Pharma Inc. | Milled particles |
| US6635638B2 (en) | 2000-05-17 | 2003-10-21 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted pyrrolopyridinone derivatives useful as phosphodiesterase inhibitors |
| US6638937B2 (en) | 1998-07-06 | 2003-10-28 | Bristol-Myers Squibb Co. | Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists |
| US6642244B2 (en) | 2001-03-16 | 2003-11-04 | Bristol-Myers Squibb Co. | Pyrazolopyridopyrimidine inhibitors of cGMP phosphodiesterase |
| US6642274B1 (en) | 1999-09-09 | 2003-11-04 | Gary W. Neal | Methods and compositions for preventing and treating prostate disorders |
| US6645466B1 (en) | 1998-11-13 | 2003-11-11 | Jago Research Ag | Dry powder for inhalation |
| US6645954B2 (en) | 1998-04-09 | 2003-11-11 | Multimed Limited | Compositions comprising ethisterone or its derivatives |
| US6645965B1 (en) | 1998-06-19 | 2003-11-11 | Nicox S.A. | Nitrate salts of antihypertensive medicines |
| US6644309B2 (en) | 2001-01-12 | 2003-11-11 | Becton, Dickinson And Company | Medicament respiratory delivery device and method |
| US6649606B1 (en) | 2001-11-09 | 2003-11-18 | Bristol-Myers Squibb Co. | Tetrahydroisoquinoline analogs as modulators of chemokine receptor activity |
| US6650943B1 (en) | 2000-04-07 | 2003-11-18 | Advanced Bionics Corporation | Fully implantable neurostimulator for cavernous nerve stimulation as a therapy for erectile dysfunction and other sexual dysfunction |
| US6656452B1 (en) | 1997-10-21 | 2003-12-02 | The General Hospital Corporation | Use of inhaled NO as anti-inflammatory agent |
| US6656385B2 (en) | 2001-02-21 | 2003-12-02 | The Procter & Gamble Company | Functionalized cubic liquid crystalline phase materials and methods for their preparation and use |
| US6656935B2 (en) | 1999-09-16 | 2003-12-02 | Tanabe Seiyaku Co., Ltd. | Aromatic nitrogen-containing 6-membered cyclic compounds |
| US6660756B2 (en) | 2001-03-28 | 2003-12-09 | Pfizer Inc. | N-phenpropylcyclopentyl-substituted glutaramide derivatives as inhibitors of neutral endopeptidase |
| US6667060B1 (en) | 1999-03-31 | 2003-12-23 | Janssen Pharmaceutica N.V. | Pregelatinized starch in a controlled release formulation |
| US6667398B2 (en) | 2000-06-22 | 2003-12-23 | Pfizer Inc | Process for the preparation of pyrazolopyrimidinones |
| US6669961B2 (en) | 2000-08-15 | 2003-12-30 | Board Of Trustees Of University Of Illinois | Microparticles |
| US6670386B2 (en) | 2001-07-31 | 2003-12-30 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US6673987B1 (en) | 2000-03-24 | 2004-01-06 | Geron Corporation | Strategy for maintaining pregnancy |
| US6673841B2 (en) | 2001-12-20 | 2004-01-06 | Whan In Pharm. Co., Ltd. | Alprostadil alkyl ester-containing composition for external application |
| US6673778B1 (en) | 1996-02-23 | 2004-01-06 | The Board Of Regents Of The University Of Nebraska | Enzyme inhibitors for metabolic redirection |
| US6682716B2 (en) | 2001-06-05 | 2004-01-27 | Alexza Molecular Delivery Corporation | Delivery of aerosols containing small particles through an inhalation route |
| US6683080B2 (en) | 2001-02-02 | 2004-01-27 | Pfizer Inc. | Treatment of diabetes mellitus |
| US6686349B2 (en) | 2001-11-14 | 2004-02-03 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted tetracyclic pyrroloquinolone derivatives useful as phosphodiesterase inhibitors |
| US6689338B2 (en) | 2000-06-01 | 2004-02-10 | The Board Of Regents For Oklahoma State University | Bioconjugates of nanoparticles as radiopharmaceuticals |
| US6689118B2 (en) | 1999-10-14 | 2004-02-10 | Becton Dickinson And Company | Method of intradermally injecting substances |
| US6696495B2 (en) | 1998-12-02 | 2004-02-24 | Snowden Pharmaceuticals, Llc | Treatment of disorders secondary to organic impairments |
| US6706283B1 (en) | 1999-02-10 | 2004-03-16 | Pfizer Inc | Controlled release by extrusion of solid amorphous dispersions of drugs |
| US6706720B2 (en) | 1999-12-06 | 2004-03-16 | Bristol-Myers Squibb Company | Heterocyclic dihydropyrimidine compounds |
| US6706691B1 (en) | 1998-07-15 | 2004-03-16 | Board Of Regents, The University Of Texas System | Immunosupportive drug sparing diet |
| US6706892B1 (en) | 1999-09-07 | 2004-03-16 | Conjuchem, Inc. | Pulmonary delivery for bioconjugation |
| US6706682B2 (en) | 1999-01-21 | 2004-03-16 | The Trustees Of Columbia University In The City Of New York | Uses of vascular endothelial growth factor in the treatment of erectile dysfunction |
| US6713487B2 (en) | 2001-03-02 | 2004-03-30 | Bristol-Myers Squibb Co. | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US6730689B2 (en) | 2001-12-04 | 2004-05-04 | Bristol-Myers Squibb Company | N-[4-(1H-imidazol-1-yl)-2-fluorophenyl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as factor Xa inhibitors |
| US6730674B2 (en) | 2001-02-28 | 2004-05-04 | Pfizer Inc | Sulfonyl pyridazinone compounds useful as aldose reductase inhibitors |
| US6730786B2 (en) | 2000-06-22 | 2004-05-04 | Pfizer Inc | Process for the preparation of pyrazolopyrimidinones |
| US6734186B1 (en) | 1999-11-08 | 2004-05-11 | Pfizer Inc. | Compounds for the treatment of female sexual dysfunction |
| US6737070B1 (en) | 2001-03-06 | 2004-05-18 | Craig N. Burkhart | Methods of increasing the efficacy of peroxides |
| US6740306B2 (en) | 2001-04-12 | 2004-05-25 | Bayer Aktiengesellschaft | Imidazotriazinone-containing compositions for nasal administration |
| US6740793B2 (en) | 2001-09-12 | 2004-05-25 | Albert Einstein College Of Medicine Of Yeshiva University | Transgenic animal having a disrupted PDE7A gene and uses thereof |
| US6743443B1 (en) | 1998-10-05 | 2004-06-01 | Eisai Co., Ltd. | Tablets immediately disintegrating in the oral cavity |
| WO2005013937A2 (en) * | 2003-07-23 | 2005-02-17 | Elan Pharma International Ltd. | Novel compositions of sildenafil free base |
| WO2007080362A1 (en) | 2006-01-14 | 2007-07-19 | Pliva Hrvatska D.O.O. | Pharmaceutically acceptable co-crystalline forms of sildenafil |
| WO2007110559A1 (en) | 2006-03-29 | 2007-10-04 | Pliva Hrvatska D.O.O. | Pharmaceutically acceptable salts and polymorphic forms of sildenafil |
| PT1633364E (en) | 2004-01-05 | 2008-05-29 | Teva Pharma | Processes for the production of sildenafil base and citrate salt |
| WO2010146407A1 (en) | 2009-06-19 | 2010-12-23 | Nanoform Hungary Ltd. | Nanostructured sildenafil base, its pharmaceutically acceptable salts and co-crystals, compositions of them, process for the preparation thereof and pharmaceutical compositions containing them |
| EP2374460A1 (en) * | 2008-12-12 | 2011-10-12 | Siegfried Rhein S.A. De C.V. | Pulsed-release sildenafil composition and method for preparing said composition |
| WO2013001516A1 (en) * | 2011-06-29 | 2013-01-03 | Ranbaxy Laboratories Limited | Multilayered dosage form |
-
2017
- 2017-03-31 WO PCT/GB2017/050921 patent/WO2017168174A1/en active Application Filing
Patent Citations (298)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3760984A (en) | 1971-09-29 | 1973-09-25 | Alza Corp | Osmotically powered agent dispensing device with filling means |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US4036227A (en) | 1973-04-25 | 1977-07-19 | Alza Corporation | Osmotic releasing device having a plurality of release rate patterns |
| US3987790A (en) | 1975-10-01 | 1976-10-26 | Alza Corporation | Osmotically driven fluid dispenser |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US4681583A (en) | 1982-12-20 | 1987-07-21 | Alza Corporation | System for dispersing drug in biological environment |
| US4578075A (en) | 1982-12-20 | 1986-03-25 | Alza Corporation | Delivery system housing a plurality of delivery devices |
| US4773907A (en) | 1982-12-20 | 1988-09-27 | Alza Corporation | Primary delivery system comprising secondary dosage form |
| US4576604A (en) | 1983-03-04 | 1986-03-18 | Alza Corporation | Osmotic system with instant drug availability |
| US4673405A (en) | 1983-03-04 | 1987-06-16 | Alza Corporation | Osmotic system with instant drug availability |
| US4693895A (en) | 1984-10-26 | 1987-09-15 | Alza Corporation | Colon delivery system |
| US4705515A (en) | 1984-10-26 | 1987-11-10 | Alza Corporation | Dosage form for administering drug of the colon |
| US4642903A (en) | 1985-03-26 | 1987-02-17 | R. P. Scherer Corporation | Freeze-dried foam dosage form |
| US4855326A (en) | 1987-04-20 | 1989-08-08 | Fuisz Pharmaceutical Ltd. | Rapidly dissoluble medicinal dosage unit and method of manufacture |
| US5178878A (en) | 1989-10-02 | 1993-01-12 | Cima Labs, Inc. | Effervescent dosage form with microparticles |
| US5229133A (en) | 1990-01-24 | 1993-07-20 | Alza Corporation | Delivery system comprising means for controlling internal pressure |
| US5250534A (en) | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| EP0463756A1 (en) | 1990-06-20 | 1992-01-02 | Pfizer Limited | Pyrazolopyrimidinone antianginal agents |
| US6291528B1 (en) | 1991-07-03 | 2001-09-18 | Nathan Earl Scott | Prostaglandin E1/F2 in combination with prostaglandin F2α for enhancing female sexual arousal |
| US5464632A (en) | 1991-07-22 | 1995-11-07 | Laboratoires Prographarm | Rapidly disintegratable multiparticular tablet |
| US5464632C1 (en) | 1991-07-22 | 2001-02-20 | Prographarm Lab | Rapidly disintegratable multiparticular tablet |
| US6268338B1 (en) | 1993-04-30 | 2001-07-31 | Merck & Co., Inc. | Cyclohexapeptidyl amine compounds |
| US6469012B1 (en) | 1993-06-09 | 2002-10-22 | Pfizer Inc | Pyrazolopyrimidinones for the treatment of impotence |
| US6143746A (en) | 1994-01-21 | 2000-11-07 | Icos Corporation | Tetracyclic cyclic GMP-specific phosphodiesterase inhibitors, process of preparation and use |
| US5576014A (en) | 1994-01-31 | 1996-11-19 | Yamanouchi Pharmaceutical Co., Ltd | Intrabuccally dissolving compressed moldings and production process thereof |
| US6395744B1 (en) | 1994-04-22 | 2002-05-28 | Queen's University At Kingston | Method and compositions for the treatment or amelioration of female sexual dysfunction |
| US5738875A (en) | 1994-10-28 | 1998-04-14 | R.P. Scherer Corporation | Process for preparing solid pharmaceutical dosage forms |
| US6166061A (en) | 1995-04-28 | 2000-12-26 | Zonagen, Inc. | Methods and formulations for modulating the human sexual response |
| US6100286A (en) | 1995-04-28 | 2000-08-08 | Zonagen, Inc. | Methods and formulations for modulating the human sexual response |
| US5981563A (en) | 1995-04-28 | 1999-11-09 | Zonagen, Inc. | Methods and formulations for modulating the human sexual response |
| US6051594A (en) | 1995-04-28 | 2000-04-18 | Zonagen, Inc. | Methods and formulations for modulating the human sexual response |
| US6482398B1 (en) | 1995-06-07 | 2002-11-19 | The Procter & Gamble Company | Transfer-resistant lip compositions |
| US6605627B2 (en) | 1995-06-07 | 2003-08-12 | The Salk Insitute For Biological Studies | Modulators of peroxisome proliferator activated receptor-gamma, and methods for the use thereof |
| US5607697A (en) | 1995-06-07 | 1997-03-04 | Cima Labs, Incorporated | Taste masking microparticles for oral dosage forms |
| US6143757A (en) | 1995-07-14 | 2000-11-07 | Icos Corporation | Chemical compounds |
| US6723345B2 (en) | 1995-09-29 | 2004-04-20 | L.A.M. Pharmaceutical Corp. | Topical drug preparations |
| US6387407B1 (en) | 1995-09-29 | 2002-05-14 | L.A.M. Pharmaceutical Corporation | Topical drug preparations |
| US6251436B1 (en) | 1995-09-29 | 2001-06-26 | L.A.M. Pharmaceutical Corporation | Drug preparations for treating sexual dysfunction |
| US6346271B1 (en) | 1995-09-29 | 2002-02-12 | L. A. M. Pharmaceutical Corporation | Topical drug preparations |
| US6469065B1 (en) | 1996-02-02 | 2002-10-22 | Nitromed, Inc. | Nitrosated and nitrosylated α-adrenergic receptor antagonist, compositions and methods of use |
| US6673778B1 (en) | 1996-02-23 | 2004-01-06 | The Board Of Regents Of The University Of Nebraska | Enzyme inhibitors for metabolic redirection |
| US6200571B1 (en) | 1996-06-10 | 2001-03-13 | Lotfi Ismail Omar | Methods and compositions for the treatment of sexual dysfunction in humans and animals |
| US5955611A (en) | 1996-06-14 | 1999-09-21 | Pfizer Inc. | Process for preparing sildenafil |
| US6066735A (en) | 1996-06-14 | 2000-05-23 | Pfizer Inc. | Process for preparing sildenafil |
| US6572880B2 (en) | 1996-10-24 | 2003-06-03 | Pharmaceutical Applications Associates Llc | Methods and transdermal compositions for pain relief |
| US6479074B2 (en) | 1996-10-24 | 2002-11-12 | Pharmaceutical Applications Associates Llc | Methods and transdermal compositions for pain relief |
| US6290986B1 (en) | 1996-10-24 | 2001-09-18 | Pharmaceutical Applications Associates, Llc | Method and composition for transdermal administration of pharmacologic agents |
| US6462044B2 (en) | 1996-11-01 | 2002-10-08 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitors, compositions and methods of use |
| US6331543B1 (en) | 1996-11-01 | 2001-12-18 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitors, compositions and methods of use |
| US5874437A (en) | 1996-11-01 | 1999-02-23 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6172068B1 (en) | 1996-11-01 | 2001-01-09 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6172060B1 (en) | 1996-11-01 | 2001-01-09 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6177428B1 (en) | 1996-11-01 | 2001-01-23 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6133272A (en) | 1996-11-01 | 2000-10-17 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| USRE37234E1 (en) | 1996-11-01 | 2001-06-19 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiestrase inhibitor compounds, compositions and their uses |
| US5958926A (en) | 1996-11-01 | 1999-09-28 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6221881B1 (en) | 1996-11-01 | 2001-04-24 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6197782B1 (en) | 1996-11-01 | 2001-03-06 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6197778B1 (en) | 1996-11-01 | 2001-03-06 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6232321B1 (en) | 1996-11-01 | 2001-05-15 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6211179B1 (en) | 1996-11-01 | 2001-04-03 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6316457B1 (en) | 1996-11-01 | 2001-11-13 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US6413496B1 (en) | 1996-12-04 | 2002-07-02 | Biogland Ireland (R&D) Limited | Pharmaceutical compositions and devices for their administration |
| US6271211B1 (en) | 1997-02-13 | 2001-08-07 | Albert Einstein College Of Medicine Of Yeshiva University | Gene therapy for regulating penile smooth muscle tone |
| US6239117B1 (en) | 1997-02-13 | 2001-05-29 | Albert Einstein College Of Medicine Of Yeshiva University | Gene therapy for regulating bladder smooth muscle tone |
| US6333354B1 (en) | 1997-02-28 | 2001-12-25 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Synergistic combination of PDE inhibitors and adenylate cyclase agonists or guanyl cyclyse agonists |
| US6013663A (en) | 1997-04-02 | 2000-01-11 | Sankyo Company, Limited | Dithiolan derivatives, their preparation and their therapeutic effect |
| US6313164B1 (en) | 1997-04-02 | 2001-11-06 | Sankyo Company, Limited | Dithiolin derivatives, their preparation and their therapeutic effect |
| US6043252A (en) | 1997-05-05 | 2000-03-28 | Icos Corporation | Carboline derivatives |
| US6589990B1 (en) | 1997-05-06 | 2003-07-08 | Panagiotis Kanakaris | Methods and compositions for misoprostol compound treatment of erectile dysfunction |
| US6500610B1 (en) | 1997-05-30 | 2002-12-31 | Cell Pathways, Inc | Methods for identifying compounds for inhibiting of neoplastic lesions, and pharmaceutical compositions containing such compounds |
| US5858694A (en) | 1997-05-30 | 1999-01-12 | Cell Pathways, Inc. | Method for identifying compounds for inhibition of cancerous lesions |
| US6610652B2 (en) | 1997-06-23 | 2003-08-26 | Queen's University At Kingston | Microdose therapy |
| US6423683B1 (en) | 1997-06-23 | 2002-07-23 | Queens University At Kingston | Microdose therapy |
| US6165975A (en) | 1997-06-23 | 2000-12-26 | Queen's University At Kingston | Microdose therapy |
| US6410595B1 (en) | 1997-07-09 | 2002-06-25 | Androsolutions, Inc. | Methods and compositions for treating male erectile dysfunction |
| US6559184B2 (en) | 1997-07-09 | 2003-05-06 | Androsolutions, Inc. | Methods and compositions for treating male erectile dysfunction |
| US6414027B1 (en) | 1997-07-09 | 2002-07-02 | Androsolutions, Inc. | Methods and compositions for treating male erectile dysfunction |
| US6476074B1 (en) | 1997-07-10 | 2002-11-05 | Synphora Ab | Method and composition for treatment of erectile dysfunction |
| US6593369B2 (en) | 1997-10-20 | 2003-07-15 | Vivus, Inc. | Methods, compositions, and kits for enhancing female sexual desire and responsiveness |
| US6656452B1 (en) | 1997-10-21 | 2003-12-02 | The General Hospital Corporation | Use of inhaled NO as anti-inflammatory agent |
| US6593313B2 (en) | 1997-10-28 | 2003-07-15 | Vivus, Inc. | Co-administration of a prostaglandin and an androgenic agent in the treatment of female sexual dysfunction |
| US6306841B1 (en) | 1997-10-28 | 2001-10-23 | Asivi, Llc | Treatment of female sexual dysfunction |
| US6037346A (en) | 1997-10-28 | 2000-03-14 | Vivus, Inc. | Local administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6403597B1 (en) | 1997-10-28 | 2002-06-11 | Vivus, Inc. | Administration of phosphodiesterase inhibitors for the treatment of premature ejaculation |
| US6127363A (en) | 1997-10-28 | 2000-10-03 | Vivus, Inc. | Local administration of Type IV phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6472434B1 (en) | 1997-10-28 | 2002-10-29 | Vivus, Inc. | Method for minimizing excess collagen deposition |
| US6156753A (en) | 1997-10-28 | 2000-12-05 | Vivus, Inc. | Local administration of type III phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6548490B1 (en) | 1997-10-28 | 2003-04-15 | Vivus, Inc. | Transmucosal administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6469016B1 (en) | 1997-10-28 | 2002-10-22 | Vivus, Inc. | Treatment of female sexual dysfunction using phosphodiesterase inhibitors |
| US6294550B1 (en) | 1997-10-28 | 2001-09-25 | Asivi, Llc | Treatment of female sexual dysfunction |
| US6472425B1 (en) | 1997-10-31 | 2002-10-29 | Nitromed, Inc. | Methods for treating female sexual dysfunctions |
| US6362178B1 (en) | 1997-11-12 | 2002-03-26 | Bayer Aktiengesellschaft | 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| US6566360B1 (en) | 1997-11-12 | 2003-05-20 | Bayer Aktiengesellschaft | 2-phenyl substituted imidatriazinones as phosphodiesterase inhibitors |
| US6221402B1 (en) | 1997-11-20 | 2001-04-24 | Pfizer Inc. | Rapidly releasing and taste-masking pharmaceutical dosage form |
| US6476021B1 (en) | 1997-11-28 | 2002-11-05 | Mochida Pharmaceutical Co., Ltd. | Compounds having cGMP-PDE inhibitory effect |
| US6342251B1 (en) | 1997-12-02 | 2002-01-29 | West Pharmaceutical Services Drug Delivery & Clinical Research Centre Limited | Compositions for nasal administration |
| US6368640B1 (en) | 1998-04-03 | 2002-04-09 | The Daily Wellness Company | Method and composition for improving sexual fitness |
| US6544563B2 (en) | 1998-04-03 | 2003-04-08 | The Daily Wellness Company | Method and composition for improving sexual fitness |
| US6645954B2 (en) | 1998-04-09 | 2003-11-11 | Multimed Limited | Compositions comprising ethisterone or its derivatives |
| US6541487B1 (en) | 1998-05-01 | 2003-04-01 | R.T. Alamo Ventures I, Llc | PDE III inhibitors for treating sexual dysfunction |
| US6258373B1 (en) | 1998-05-01 | 2001-07-10 | Neal R. Cutler | Treatment of sexual dysfunction in certain patient groups |
| US6132757A (en) | 1998-05-01 | 2000-10-17 | Neal R. Cutler | Treatment of sexual dysfunction in certain patient groups |
| US6241752B1 (en) | 1998-05-08 | 2001-06-05 | Inventis | Method of treating for impotence and apparatus particularly useful in such method |
| US6204383B1 (en) | 1998-05-15 | 2001-03-20 | Torcan Chemical Ltd. | Processes for preparing sildenafil |
| US6124461A (en) | 1998-05-26 | 2000-09-26 | Saint Louis University, Health Services Center, Research Administration | Compounds, compositions, and methods for treating erectile dysfunction |
| US6365590B1 (en) | 1998-05-26 | 2002-04-02 | Saint Louis University | Compounds, compositions and methods for treating erectile dysfunction |
| US6277884B1 (en) | 1998-06-01 | 2001-08-21 | Nitromed, Inc. | Treatment of sexual dysfunction with N-hydroxyguanidine compounds |
| US6436997B1 (en) | 1998-06-01 | 2002-08-20 | Nitromed, Inc. | Endogenous nitric oxide synthesis under conditions of low oxygen tension |
| US6087368A (en) | 1998-06-08 | 2000-07-11 | Bristol-Myers Squibb Company | Quinazolinone inhibitors of cGMP phosphodiesterase |
| US6410548B2 (en) | 1998-06-11 | 2002-06-25 | Merck & Co., Inc. | Spiropiperidine derivatives as melanocortin receptor agonists |
| US6294534B1 (en) | 1998-06-11 | 2001-09-25 | Merck & Co., Inc. | Spiropiperidine derivatives as melanocortin receptor agonists |
| US6645965B1 (en) | 1998-06-19 | 2003-11-11 | Nicox S.A. | Nitrate salts of antihypertensive medicines |
| US6266560B1 (en) | 1998-06-19 | 2001-07-24 | Genetronics, Inc. | Electrically assisted transdermal method and apparatus for the treatment of erectile dysfunction |
| US6596733B2 (en) | 1998-06-23 | 2003-07-22 | Medinox, Inc. | Use of nitric oxide scavengers to treat side effects caused by therapeutic administration of sources of nitric oxide |
| US6265420B1 (en) | 1998-06-23 | 2001-07-24 | Medinox, Inc. | Use of nitric oxide scavengers to treat side effects caused by therapeutic administration of sources of nitric oxide |
| US6200591B1 (en) | 1998-06-25 | 2001-03-13 | Anwar A. Hussain | Method of administration of sildenafil to produce instantaneous response for the treatment of erectile dysfunction |
| US6638937B2 (en) | 1998-07-06 | 2003-10-28 | Bristol-Myers Squibb Co. | Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists |
| US6007824A (en) | 1998-07-09 | 1999-12-28 | Duckett; Melvin J. | Natural composition and method for the treatment of sexual dysfunction |
| US6706691B1 (en) | 1998-07-15 | 2004-03-16 | Board Of Regents, The University Of Texas System | Immunosupportive drug sparing diet |
| US6251428B1 (en) | 1998-07-24 | 2001-06-26 | Seo Hong Yoo | Preparation of aqueous clear solution dosage forms with bile acids |
| US6585958B1 (en) | 1998-07-24 | 2003-07-01 | Jago Research Ag | Medicinal aerosol formulations |
| US6284763B1 (en) | 1998-08-26 | 2001-09-04 | Queen's University At Kingston | Methods for remodeling neuronal and cardiovascular pathways |
| US6458797B1 (en) | 1998-08-26 | 2002-10-01 | Queen's University Of Kingston | Methods for remodeling neuronal and cardiovascular pathways |
| US6077841A (en) | 1998-09-04 | 2000-06-20 | Janssen Pharmaceutica, N.V. | 5-heterocyclyl pyrazolo[4,3-d]pyrimidin-7-ones for the treatment of male erectile dysfunction |
| US6514536B2 (en) | 1998-09-08 | 2003-02-04 | L.A.M. Pharmaceutical Corporation | Drug preparations for treating sexual dysfunction |
| US6462047B1 (en) | 1998-09-16 | 2002-10-08 | Icos Corporation | Carboline derivatives as cGMP phosphodiesterase inhibitors |
| US6326379B1 (en) | 1998-09-16 | 2001-12-04 | Bristol-Myers Squibb Co. | Fused pyridine inhibitors of cGMP phosphodiesterase |
| US6696072B1 (en) | 1998-09-17 | 2004-02-24 | Zonagen, Inc. | Methods for treatment of male erectile dysfunction |
| US6482426B1 (en) | 1998-09-17 | 2002-11-19 | Zonagen, Inc. | Compositions for the treatment of male erectile dysfunction |
| US6743443B1 (en) | 1998-10-05 | 2004-06-01 | Eisai Co., Ltd. | Tablets immediately disintegrating in the oral cavity |
| US6194433B1 (en) | 1998-10-05 | 2001-02-27 | Neal R. Cutler | Sexual dysfunction in females |
| US6207829B1 (en) | 1998-10-12 | 2001-03-27 | Pfizer Inc. | Process for preparation of pyrazolo[4,3-d]pyrimidin-7-ones and intermediates thereof |
| US6200771B1 (en) | 1998-10-15 | 2001-03-13 | Cell Pathways, Inc. | Method of using a novel phosphodiesterase in pharmaceutical screeing to identify compounds for treatment of neoplasia |
| WO2000024383A1 (en) * | 1998-10-23 | 2000-05-04 | Pfizer Research And Development Company, N.V./S.A. | Controlled-release pharmaceutical formulations containing a cgmp pde-5 inhibitor |
| US6485747B1 (en) | 1998-10-30 | 2002-11-26 | Monsanto Company | Coated active tablet(s) |
| US6583147B1 (en) | 1998-11-11 | 2003-06-24 | Dong A Pharm Co., Ltd. | Pyrazolopyrimidinone derivatives for the treatment of impotence |
| US6235782B1 (en) | 1998-11-12 | 2001-05-22 | Rifat Pamukcu | Method for treating a patient with neoplasia by treatment with a platinum coordination complex |
| US6472420B2 (en) | 1998-11-12 | 2002-10-29 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a paclitaxel derivative |
| US6365627B2 (en) | 1998-11-12 | 2002-04-02 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a paclitaxel derivative |
| US6235776B1 (en) | 1998-11-12 | 2001-05-22 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a paclitaxel derivative |
| US6359002B2 (en) | 1998-11-12 | 2002-03-19 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a platinum coordination complex |
| US6645466B1 (en) | 1998-11-13 | 2003-11-11 | Jago Research Ag | Dry powder for inhalation |
| US6696495B2 (en) | 1998-12-02 | 2004-02-24 | Snowden Pharmaceuticals, Llc | Treatment of disorders secondary to organic impairments |
| US6323242B1 (en) | 1998-12-02 | 2001-11-27 | Peter Sterling Mueller | Treatment of disorders secondary to organic impairments |
| US6184231B1 (en) | 1998-12-04 | 2001-02-06 | Bristol-Myers Squibb | 3-substituted-4-arylquinolin-2-one derivatives as potassium channel modulators |
| US6395736B1 (en) | 1998-12-14 | 2002-05-28 | Cellegy Pharmaceuticals, Inc. | Compositions and methods for the treatment of anorectal disorders |
| US6391869B1 (en) | 1998-12-14 | 2002-05-21 | Cellergy Pharmaceuticals, Inc. | Compositions and methods for the treatment of anorectal disorders |
| US6627632B2 (en) | 1998-12-14 | 2003-09-30 | Cellegy Pharmaceuticals, Inc. | Compositions and methods for the treatment of anorectal disorders |
| US6592850B2 (en) | 1998-12-15 | 2003-07-15 | Wm. Wrigley Jr. Company | Sildenafil citrate chewing gum formulations and methods of using the same |
| US6627234B1 (en) | 1998-12-15 | 2003-09-30 | Wm. Wrigley Jr. Company | Method of producing active agent coated chewing gum products |
| US6291471B1 (en) | 1998-12-17 | 2001-09-18 | Abb Holdings, Inc. | Use of apomorphine for the treatment of organic erectile dysfunction in males |
| US6562868B1 (en) | 1999-01-08 | 2003-05-13 | Synphora Ab | Method for treatment of female sexual dysfunction |
| US6552024B1 (en) | 1999-01-21 | 2003-04-22 | Lavipharm Laboratories Inc. | Compositions and methods for mucosal delivery |
| US6706682B2 (en) | 1999-01-21 | 2004-03-16 | The Trustees Of Columbia University In The City Of New York | Uses of vascular endothelial growth factor in the treatment of erectile dysfunction |
| US6417208B1 (en) | 1999-02-05 | 2002-07-09 | Albert Einstein College Of Medicine Of Yeshiva University | Method of identification of inhibitors of PDE1C |
| US6706283B1 (en) | 1999-02-10 | 2004-03-16 | Pfizer Inc | Controlled release by extrusion of solid amorphous dispersions of drugs |
| US6294192B1 (en) | 1999-02-26 | 2001-09-25 | Lipocine, Inc. | Triglyceride-free compositions and methods for improved delivery of hydrophobic therapeutic agents |
| US6451339B2 (en) | 1999-02-26 | 2002-09-17 | Lipocine, Inc. | Compositions and methods for improved delivery of hydrophobic agents |
| US6187790B1 (en) | 1999-03-04 | 2001-02-13 | Neal R. Cutler | Use of cilostazol for treatment of sexual dysfunction |
| US6087362A (en) | 1999-03-16 | 2000-07-11 | Pentech Pharmaceuticals, Inc. | Apomorphine and sildenafil composition |
| US6376554B1 (en) | 1999-03-19 | 2002-04-23 | Knoll Pharmaceutical Company | Method of treating sexual dysfunction |
| US6316438B1 (en) | 1999-03-22 | 2001-11-13 | Bristol-Myers Squibb Co. | Fused pyridopyridazine inhibitors of cGMP phosphodiesterase |
| US6482948B1 (en) | 1999-03-30 | 2002-11-19 | Nippon Soda Co., Ltd. | Thienopyrimidine compounds and salts thereof and process for the preparation of the same |
| US6667060B1 (en) | 1999-03-31 | 2003-12-23 | Janssen Pharmaceutica N.V. | Pregelatinized starch in a controlled release formulation |
| US6102849A (en) | 1999-04-03 | 2000-08-15 | Hakac; John R. | Non-surgical penile prosthesis |
| US6383471B1 (en) | 1999-04-06 | 2002-05-07 | Lipocine, Inc. | Compositions and methods for improved delivery of ionizable hydrophobic therapeutic agents |
| US6531114B1 (en) | 1999-04-06 | 2003-03-11 | Wm. Wrigley Jr. Company | Sildenafil citrate chewing gum formulations and methods of using the same |
| US6214849B1 (en) | 1999-04-29 | 2001-04-10 | Lupin Laboratories Limited | Use of nicorandil in treatment of sexual dysfunction or for enhancement of sexual function in mammals including humans |
| US6451807B1 (en) | 1999-04-30 | 2002-09-17 | Lilly Icos, Llc. | Methods of treating sexual dysfunction in an individual suffering from a retinal disease, class 1 congestive heart failure, or myocardial infarction using a PDE5 inhibitor |
| US6613768B1 (en) | 1999-04-30 | 2003-09-02 | Lilly Icos Llc | Treatment of female arousal disorder |
| US6632419B2 (en) | 1999-05-04 | 2003-10-14 | Aradigm Corporation | Increasing libido in humans via acute testosterone administration |
| US6428769B1 (en) | 1999-05-04 | 2002-08-06 | Aradigm Corporation | Acute testosterone administration |
| US6303606B1 (en) | 1999-05-07 | 2001-10-16 | Recordati, S.A., Chemical And Pharmaceutical Company | Use of selective antagonists of the α1B-adrenergic receptor for improvement of sexual dysfunction |
| US6417207B1 (en) | 1999-05-12 | 2002-07-09 | Nitromed, Inc. | Nitrosated and nitrosylated potassium channel activators, compositions and methods of use |
| US6693122B2 (en) | 1999-05-12 | 2004-02-17 | Nitromed, Inc. | Nitrosated and nitrosylated potassium channel activators, compositions and methods of use |
| US6395300B1 (en) | 1999-05-27 | 2002-05-28 | Acusphere, Inc. | Porous drug matrices and methods of manufacture thereof |
| US6645528B1 (en) | 1999-05-27 | 2003-11-11 | Acusphere, Inc. | Porous drug matrices and methods of manufacture thereof |
| US6350760B1 (en) | 1999-06-04 | 2002-02-26 | Merck & Co., Inc. | Substituted piperidines as melanocortin-4 receptor agonists |
| US6242444B1 (en) | 1999-06-04 | 2001-06-05 | The Jordanian Pharmaceutical Manufacturing And Medical Equipment Co., Ltd. | Compounds and pharmaceutical compositions containing the same |
| US6579968B1 (en) | 1999-06-29 | 2003-06-17 | Palatin Technologies, Inc. | Compositions and methods for treatment of sexual dysfunction |
| US6267985B1 (en) | 1999-06-30 | 2001-07-31 | Lipocine Inc. | Clear oil-containing pharmaceutical compositions |
| US6303135B1 (en) | 1999-07-08 | 2001-10-16 | Neal R. Cutler | Use of quinolines and quinolones to treat male erectile dysfunction |
| US6548087B1 (en) | 1999-07-12 | 2003-04-15 | Frances B. Kent | Nutritional supplement |
| US6130053A (en) | 1999-08-03 | 2000-10-10 | Cell Pathways, Inc. | Method for selecting compounds for inhibition of neoplastic lesions |
| US6706892B1 (en) | 1999-09-07 | 2004-03-16 | Conjuchem, Inc. | Pulmonary delivery for bioconjugation |
| US6642274B1 (en) | 1999-09-09 | 2003-11-04 | Gary W. Neal | Methods and compositions for preventing and treating prostate disorders |
| US6380267B1 (en) | 1999-09-13 | 2002-04-30 | David M. Swope | Composition and method for decreasing neurologic symptomatology |
| US6656935B2 (en) | 1999-09-16 | 2003-12-02 | Tanabe Seiyaku Co., Ltd. | Aromatic nitrogen-containing 6-membered cyclic compounds |
| US6075028A (en) | 1999-09-23 | 2000-06-13 | Graham; Richard | Method of treating Tourette's syndrome and related CNS disorders |
| US6576653B2 (en) | 1999-09-30 | 2003-06-10 | Pfizer Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
| US6399601B1 (en) | 1999-09-30 | 2002-06-04 | Pfizer Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
| US6436944B1 (en) | 1999-09-30 | 2002-08-20 | Pfizer Inc. | Combination effective for the treatment of impotence |
| US6569123B2 (en) | 1999-10-14 | 2003-05-27 | Becton, Dickinson And Company | Prefillable intradermal injector |
| US6569143B2 (en) | 1999-10-14 | 2003-05-27 | Becton, Dickinson And Company | Method of intradermally injecting substances |
| US6689118B2 (en) | 1999-10-14 | 2004-02-10 | Becton Dickinson And Company | Method of intradermally injecting substances |
| US6622721B2 (en) | 1999-10-14 | 2003-09-23 | Becton, Dickinson And Company | Drug delivery system including holder and drug container |
| US6734186B1 (en) | 1999-11-08 | 2004-05-11 | Pfizer Inc. | Compounds for the treatment of female sexual dysfunction |
| US6211156B1 (en) | 1999-11-10 | 2001-04-03 | Asta Medica A.G. | Peptides for treatment of erectile dysfunction |
| US6548544B1 (en) | 1999-11-18 | 2003-04-15 | The National University Of Singapore | Method of treatment of priapism without raising systemic blood pressure |
| US6548044B1 (en) | 1999-11-22 | 2003-04-15 | Epix Medical, Inc. | Imaging sexual response |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6569463B2 (en) | 1999-11-23 | 2003-05-27 | Lipocine, Inc. | Solid carriers for improved delivery of hydrophobic active ingredients in pharmaceutical compositions |
| US6706720B2 (en) | 1999-12-06 | 2004-03-16 | Bristol-Myers Squibb Company | Heterocyclic dihydropyrimidine compounds |
| US6512002B2 (en) | 2000-01-12 | 2003-01-28 | Pfizer Inc. | Methods of treatment for premature ejaculation in a male |
| US6586478B2 (en) | 2000-02-22 | 2003-07-01 | Cellegy Canada | Methods and compositions for improving sleep |
| US6555547B1 (en) | 2000-02-28 | 2003-04-29 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a vinca alkaloid derivative |
| US6569638B1 (en) | 2000-03-03 | 2003-05-27 | Cell Pathways, Inc | Method for screening compounds for the treatment of neoplasia |
| US6465465B1 (en) | 2000-03-14 | 2002-10-15 | Arzneimittelwerk Dresden Gmbh | Process for the treatment of erectile dysfunction and product therefor |
| US6458790B2 (en) | 2000-03-23 | 2002-10-01 | Merck & Co., Inc. | Substituted piperidines as melanocortin receptor agonists |
| US6476037B1 (en) | 2000-03-23 | 2002-11-05 | The Regents Of The University Of California | L-arginine and phosphodiesterase (PDE) inhibitor synergism |
| US6673987B1 (en) | 2000-03-24 | 2004-01-06 | Geron Corporation | Strategy for maintaining pregnancy |
| US6436684B1 (en) | 2000-03-27 | 2002-08-20 | Applera Corporation | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6448293B1 (en) | 2000-03-31 | 2002-09-10 | Pfizer Inc. | Diphenyl ether compounds useful in therapy |
| US6455572B1 (en) | 2000-04-07 | 2002-09-24 | Pfizer Inc. | Estrogen agonist/antagonist metabolites |
| US6650943B1 (en) | 2000-04-07 | 2003-11-18 | Advanced Bionics Corporation | Fully implantable neurostimulator for cavernous nerve stimulation as a therapy for erectile dysfunction and other sexual dysfunction |
| US6492371B2 (en) | 2000-04-19 | 2002-12-10 | H H. Roylance | Use of cyclic GMP-specific phosphodiesterase inhibitors for treatment of Parkinson's Disease |
| US6271228B1 (en) | 2000-04-28 | 2001-08-07 | Pfizer Inc. | Blood pressure stabilization during hemodialysis |
| US6604698B2 (en) | 2000-05-10 | 2003-08-12 | Skyepharma Canada, Inc. | Media milling |
| US6469024B2 (en) | 2000-05-11 | 2002-10-22 | Bristol-Myers Squibb Company | Tetrahydroisoquinoline analogs useful as growth hormone secretagogues |
| US6492358B2 (en) | 2000-05-17 | 2002-12-10 | Ortho-Mcneil Pharmaceutical, Inc. | β-carboline derivatives useful as inhibitors of phosphodiesterase |
| US6635638B2 (en) | 2000-05-17 | 2003-10-21 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted pyrrolopyridinone derivatives useful as phosphodiesterase inhibitors |
| US6499984B1 (en) | 2000-05-22 | 2002-12-31 | Warner-Lambert Company | Continuous production of pharmaceutical granulation |
| US6565851B2 (en) | 2000-05-26 | 2003-05-20 | Horphag Research Limited | Relieving symptoms of erectile dysfunction with proanthocyanidins |
| US6376509B2 (en) | 2000-05-30 | 2002-04-23 | Merck & Co., Inc. | Melanocortin receptor agonists |
| US6477410B1 (en) | 2000-05-31 | 2002-11-05 | Biophoretic Therapeutic Systems, Llc | Electrokinetic delivery of medicaments |
| US6735470B2 (en) | 2000-05-31 | 2004-05-11 | Biophoretic Therapeutic Systems, Llc | Electrokinetic delivery of medicaments |
| US6689338B2 (en) | 2000-06-01 | 2004-02-10 | The Board Of Regents For Oklahoma State University | Bioconjugates of nanoparticles as radiopharmaceuticals |
| US6544981B2 (en) | 2000-06-09 | 2003-04-08 | Bristol-Myers Squibb Company | Lactam inhibitors of factor Xa and method |
| US6426084B1 (en) | 2000-06-19 | 2002-07-30 | Neal R. Cutler | Treatment of sexual dysfunction in certain patient groups |
| US6667398B2 (en) | 2000-06-22 | 2003-12-23 | Pfizer Inc | Process for the preparation of pyrazolopyrimidinones |
| US6730786B2 (en) | 2000-06-22 | 2004-05-04 | Pfizer Inc | Process for the preparation of pyrazolopyrimidinones |
| US6500440B1 (en) | 2000-06-23 | 2002-12-31 | Whan In Pharm Co., Ltd. | Topical preparation of alprostadil for the treatment of erectile dysfunction |
| US6413968B1 (en) | 2000-06-30 | 2002-07-02 | O'donnell, Jr. Francis E. | Treatment of myasthenia gravis by cyclic nucleotide phosphodiesterase inhibition |
| US6541638B2 (en) | 2000-07-19 | 2003-04-01 | Hoffman-La Roche Inc. | Pyrrolidine derivatives |
| US6699991B2 (en) | 2000-07-28 | 2004-03-02 | Pfizer Inc. | Process for the preparation of pyrazoles |
| US6407259B1 (en) | 2000-07-28 | 2002-06-18 | Pfizer Inc. | Process for the preparation of pyrazoles |
| US6511973B2 (en) | 2000-08-02 | 2003-01-28 | Bristol-Myers Squibb Co. | Lactam inhibitors of FXa and method |
| US6399579B1 (en) | 2000-08-15 | 2002-06-04 | Hauser, Inc. | Compositions comprising icariside I and anhydroicaritin and methods for making the same |
| US6669961B2 (en) | 2000-08-15 | 2003-12-30 | Board Of Trustees Of University Of Illinois | Microparticles |
| US6634576B2 (en) | 2000-08-31 | 2003-10-21 | Rtp Pharma Inc. | Milled particles |
| US6630504B2 (en) | 2000-08-31 | 2003-10-07 | Pfizer Inc. | Phenoxyphenylheterocyclyl derivatives as SSRIs |
| US6610747B2 (en) | 2000-08-31 | 2003-08-26 | Pfizer Inc. | Phenoxybenzylamine derivatives as SSRIs |
| US6455654B1 (en) | 2000-09-01 | 2002-09-24 | Samsung Electronics Co., Ltd. | Water-soluble polymeric adhesion promoter and production method |
| US6576644B2 (en) | 2000-09-06 | 2003-06-10 | Bristol-Myers Squibb Co. | Quinoline inhibitors of cGMP phosphodiesterase |
| US6403658B1 (en) | 2000-09-21 | 2002-06-11 | Shaina Toppo | Genital vasodilator |
| US6613344B2 (en) | 2000-09-28 | 2003-09-02 | Nanocyte Inc. | Methods utilizing stinging cells/capsules |
| US6531297B2 (en) | 2000-10-20 | 2003-03-11 | Applera Corporation | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6548508B2 (en) | 2000-10-20 | 2003-04-15 | Pfizer, Inc. | Use of PDE V inhibitors for improved fecundity in mammals |
| US6743799B2 (en) | 2000-10-20 | 2004-06-01 | Pfizer Inc. | Use of PDE V inhibitors for improved fecundity in mammals |
| US6635274B1 (en) | 2000-10-27 | 2003-10-21 | Biochemics, Inc. | Solution-based transdermal drug delivery system |
| US6528521B2 (en) | 2000-11-15 | 2003-03-04 | Tap Pharmaceutical Products, Inc. | Treatment of anti-depression drug-induced sexual dysfunction with apomorphine |
| US6730505B2 (en) | 2000-11-27 | 2004-05-04 | Applera Corporation | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6420150B1 (en) | 2000-11-27 | 2002-07-16 | Pe Corporation (Ny) | Isolated human drug-metabolizing proteins, nucleic acid molecules encoding human drug-metabolizing proteins, and uses thereof |
| US6573285B2 (en) | 2000-12-21 | 2003-06-03 | Bristol-Myers Squibb Co. | Method for preventing or treating pain by administering an endothelin antagonist |
| US6497885B2 (en) | 2000-12-22 | 2002-12-24 | The Daily Wellness Company | Method and composition for improving fertility health in female and male animals and humans |
| US6644309B2 (en) | 2001-01-12 | 2003-11-11 | Becton, Dickinson And Company | Medicament respiratory delivery device and method |
| US6443152B1 (en) | 2001-01-12 | 2002-09-03 | Becton Dickinson And Company | Medicament respiratory delivery device |
| US6562838B2 (en) | 2001-01-26 | 2003-05-13 | R. T. Alamo Ventures I, L.L.C. | Treatment of cardiovascular disease with quinolinone enantiomers |
| US6458804B1 (en) | 2001-01-26 | 2002-10-01 | R.T. Alamo Venturesi, Llc | Methods for the treatment of central nervous system disorders in certain patient groups |
| US6451813B1 (en) | 2001-01-26 | 2002-09-17 | R. T. Alamo Ventures I, Llc | Treatment of gastroparesis in certain patient groups |
| US6683080B2 (en) | 2001-02-02 | 2004-01-27 | Pfizer Inc. | Treatment of diabetes mellitus |
| US6656385B2 (en) | 2001-02-21 | 2003-12-02 | The Procter & Gamble Company | Functionalized cubic liquid crystalline phase materials and methods for their preparation and use |
| US6730674B2 (en) | 2001-02-28 | 2004-05-04 | Pfizer Inc | Sulfonyl pyridazinone compounds useful as aldose reductase inhibitors |
| US6713487B2 (en) | 2001-03-02 | 2004-03-30 | Bristol-Myers Squibb Co. | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US6737070B1 (en) | 2001-03-06 | 2004-05-18 | Craig N. Burkhart | Methods of increasing the efficacy of peroxides |
| US6642244B2 (en) | 2001-03-16 | 2003-11-04 | Bristol-Myers Squibb Co. | Pyrazolopyridopyrimidine inhibitors of cGMP phosphodiesterase |
| US6383789B1 (en) | 2001-03-22 | 2002-05-07 | Pe Corporation (Ny) | Isolated human UDP-glycosyltransferase, nucleic acid molecules encoding human UDP-glycosyltransferase, and uses thereof |
| US6713295B2 (en) | 2001-03-22 | 2004-03-30 | Applera Corporation | Isolated human UDP-glycosyltransferase proteins |
| US6338862B1 (en) | 2001-03-26 | 2002-01-15 | Sarfaraz K Niazi | Composition and method of use in treating sexual dysfunction using cGMP-specific phosphodiesterase type 5 inhibitors |
| US6660756B2 (en) | 2001-03-28 | 2003-12-09 | Pfizer Inc. | N-phenpropylcyclopentyl-substituted glutaramide derivatives as inhibitors of neutral endopeptidase |
| US6579879B2 (en) | 2001-03-30 | 2003-06-17 | Pfizer Inc | Pyridazinone aldose reductase inhibitors |
| US6740306B2 (en) | 2001-04-12 | 2004-05-25 | Bayer Aktiengesellschaft | Imidazotriazinone-containing compositions for nasal administration |
| US6596900B2 (en) | 2001-04-19 | 2003-07-22 | Pfizer Inc | Fused bicyclic or tricyclic amino acids |
| US6455702B1 (en) | 2001-05-16 | 2002-09-24 | Aims Fine Chemicals, Inc. | Process for the production of N,N-carbonyl diimidazole |
| US6682716B2 (en) | 2001-06-05 | 2004-01-27 | Alexza Molecular Delivery Corporation | Delivery of aerosols containing small particles through an inhalation route |
| WO2002102802A1 (en) | 2001-06-14 | 2002-12-27 | Korea Institute Of Science And Technology | Novel pyrazolopyrimidinethione derivatives. preparation methods thereof and their use as therapeutics for erectile dysfunction |
| US20030044457A1 (en) * | 2001-07-17 | 2003-03-06 | Joaquina Faour | Drug delivery device containing oseltamivir and an H1 antagonist |
| US6670386B2 (en) | 2001-07-31 | 2003-12-30 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US6479493B1 (en) | 2001-08-23 | 2002-11-12 | Cell Pathways, Inc. | Methods for treatment of type I diabetes |
| US6465494B1 (en) | 2001-08-24 | 2002-10-15 | Cell Pathways, Inc. | Methods for treatment of cystic fibrosis |
| US6740793B2 (en) | 2001-09-12 | 2004-05-25 | Albert Einstein College Of Medicine Of Yeshiva University | Transgenic animal having a disrupted PDE7A gene and uses thereof |
| US6444237B1 (en) | 2001-09-13 | 2002-09-03 | Pamela A. Heleen | Herbal composition for enhancing sexual response |
| US6624138B1 (en) | 2001-09-27 | 2003-09-23 | Gp Medical | Drug-loaded biological material chemically treated with genipin |
| US6649606B1 (en) | 2001-11-09 | 2003-11-18 | Bristol-Myers Squibb Co. | Tetrahydroisoquinoline analogs as modulators of chemokine receptor activity |
| US6686349B2 (en) | 2001-11-14 | 2004-02-03 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted tetracyclic pyrroloquinolone derivatives useful as phosphodiesterase inhibitors |
| US6730689B2 (en) | 2001-12-04 | 2004-05-04 | Bristol-Myers Squibb Company | N-[4-(1H-imidazol-1-yl)-2-fluorophenyl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as factor Xa inhibitors |
| US6673841B2 (en) | 2001-12-20 | 2004-01-06 | Whan In Pharm. Co., Ltd. | Alprostadil alkyl ester-containing composition for external application |
| US6623768B1 (en) | 2002-04-16 | 2003-09-23 | Yousry M. A. Naguib | Pharmaceutically active composition extracted from Ferula hermonis and process of its extraction |
| WO2005013937A2 (en) * | 2003-07-23 | 2005-02-17 | Elan Pharma International Ltd. | Novel compositions of sildenafil free base |
| PT1633364E (en) | 2004-01-05 | 2008-05-29 | Teva Pharma | Processes for the production of sildenafil base and citrate salt |
| WO2007080362A1 (en) | 2006-01-14 | 2007-07-19 | Pliva Hrvatska D.O.O. | Pharmaceutically acceptable co-crystalline forms of sildenafil |
| WO2007110559A1 (en) | 2006-03-29 | 2007-10-04 | Pliva Hrvatska D.O.O. | Pharmaceutically acceptable salts and polymorphic forms of sildenafil |
| EP2374460A1 (en) * | 2008-12-12 | 2011-10-12 | Siegfried Rhein S.A. De C.V. | Pulsed-release sildenafil composition and method for preparing said composition |
| WO2010146407A1 (en) | 2009-06-19 | 2010-12-23 | Nanoform Hungary Ltd. | Nanostructured sildenafil base, its pharmaceutically acceptable salts and co-crystals, compositions of them, process for the preparation thereof and pharmaceutical compositions containing them |
| RU2012101818A (en) | 2009-06-19 | 2013-07-27 | Наноформ Хунгари Лтд. | NANOSTRUCTURED BASE OF SILDENAFIL, ITS PHARMACEUTICALLY ACCEPTABLE SALTS AND CO-CRYSTALS, THEIR COMPOSITIONS, METHOD FOR PRODUCING THERE AND THEIR PHARMACEUTICAL COMPOSITIONS |
| WO2013001516A1 (en) * | 2011-06-29 | 2013-01-03 | Ranbaxy Laboratories Limited | Multilayered dosage form |
Non-Patent Citations (51)
| Title |
|---|
| "Aqueous polymeric coatings for pharmaceutical dosage forms", 1989, MARCEL DEKKER |
| "BS 5958 part 1 - Code of practice for control of undesirable static electricity", 1991, BRITISH STANDARDS INSTITUTE |
| "Dissolution technology", 1974, APS |
| "Drug Delivery Systems", 2004, CRC PRESS |
| "European Pharmacopoeia", 2001 |
| "Handbook of Powder Science & Technology", 1997, CHAP-MAN & HALL |
| "International Electrotechnical Commission", 1994 |
| "Ion exchange resins", 1981, BDH CHEMICALS LTD |
| "Ion Exchange Resins. 2nd ed.", 1958, JOHN WILEY AND SONS, INC. |
| "Pharmaceutical dosage forms: tablets", vol. 1, 1989, MARCEL DEKKKER |
| "Remington: The Science and Practice of Pharmacy. 20th ed.", 2003, LIPPINCOTT, WILLIAMS, AND WILKINS, pages: 597 |
| "Spray-drying handbook", 1991, LONGMAN SCIENTIFIC & TECHNICAL |
| "The Merck Index. 12th ed.", 1997, MERCK AND CO INC |
| "United States Pharmacopeia (USP)", 2000, pages: 1926 |
| "United States Pharmacopeia", 1999 |
| ALFONSO R GENNARO: "Remington: The Science and Practice of Pharmacy. 20th ed.", LIPPINCOTT, WILLIAMS, AND WILKINS, pages: 278 - 279 |
| AVANI P. KHRISTI ET AL, INDO AMERICAN JOURNAL OF PHARMACEUTICAL RESEARCH, vol. 5, no. 7, 2015, pages 2700 - 2708 |
| C.G. LIVERSEDGE ET AL., INT J PHARM, vol. 125, 1995, pages 309 - 313 |
| D GANDERTON: "Unit Processes in Pharmacy", vol. 7, 1968, HEINEMANN |
| DMITRIJS STEPANOVS; ANATOLY MISHNEV, ZEITSCHRIFT FUR NATURFORSCHUNG B.,, vol. 67, no. 10, 2012, pages 491 - 494 |
| DMITRIJS STEPANOVS; MARA JUREB; ANATOLY MISHNEVA, MENDELEEV COMMUN., vol. 25, 2015, pages 49 - 50 |
| FEDERAL REGISTER NOTICES, vol. 62, no. 95, pages 27115 - 27122 |
| G C COLE; J HOGAN: "Pharmaceutical coating technology", 1995, TAYLOR & FRANCIS |
| HEMMIGE S. YATHIRAJAN ET AL, ACTA CRYSTALLOGRAPHICA, vol. 61, 2005, pages 489 - 491 |
| IWONA WAWER ET AL, JOURNAL OF PARMACEUTI CAL AND BIOMEDICAL ANALYSIS, vol. 38, 2005, pages 865 - 870 |
| J R ROBINSON & V H LEE: "Controlled Drug Delivery", 1987, MARCEL DEKKER |
| K. MASTERS, SPRAY DRYING IN PRACTICE SPRAYDRYCONSULT INTERNATIONAL APS, 2002 |
| KANIG, J.PHARM SCI, vol. 53, 1964, pages 188 |
| KREUSCHNER ET AL., ACTA PHARM. TECH., vol. 23, 1980, pages 159 |
| KRISTINA MOSÉN: "Spray-drying of particles intended for inhalation by Kristina Mos'em", 2003, DANISH UNIVERSITY OF PHARMACEUTICAL SCIENCES |
| LOYD V ALLEN: "The art, science, and technology of pharmaceutical compounding", 2001, AMERICAN PHARMACEUTICAL ASSOCIATION |
| M A MCHUGH; V J KRUKONIS: "Supercritical Fluid Extraction:Principles & Practice", 1994, BUTTERWORTH-HEINEMANN |
| M E AULTON: "Pharmaceutics: the science of dosage forms", 2000 |
| M J RATHBONE; J HADGRAFT; M S ROBERTS: "Modified Release Drug Delivery Systems", 2003, MARCEL DEKKER |
| MAHMOUD M. AL OMARI ET AL, J. INCL. PHENORN. MACROCYCL. CHERN, vol. 57, 2007, pages 379 - 384 |
| MICHAEL LEVIN: "Pharmaceutical Process Scale-Up", 2003, MARCEL DEKKER |
| MIROSLAV ZEGARAC ET AL, CRYSTENGCOMM, vol. 16, 2014, pages 32 - 35 |
| MUSTAFA M. E L-ABADELAH ET AL, ZEITSCHRIFTFUR NATURFORSCHUNG B., vol. 54B, 1999, pages 1323 - 1326 |
| NOHA PATEL: "Spray-drying of pharmaceuticals for controlled release pulmonary drug delivery", UNIVERSITY OF BATH, 2000 |
| P DEASY: "Microencap-sulation and related drug processes", 1984, MARCEL DEKKER |
| PALASH SANPHUI ET AL, MOLEC ULAR PHARMACEUTICS, vol. 10, 2013, pages 4687 - 4697 |
| PALASH SANPHUI ET AL: "Salt and Cocrystals of Sildenafil with Dicarboxylic Acids: Solubility and Pharmacokinetic Advantage of the Glutarate Salt", MOLECULAR PHARMACEUTICS, vol. 10, no. 12, 2 December 2013 (2013-12-02), US, pages 4687 - 4697, XP055375732, ISSN: 1543-8384, DOI: 10.1021/mp400516b * |
| RAHUL BANERJEE ET AL, CRYSTAL GROWTH & DESIGN, vol. 6, no. 6, 2006, pages 1468 - 1478 |
| ROBERT KUNIN; R.E. KRIEGER, ION EXCHANGE RESINS, 1990 |
| ROHM; HAAS: "Ion-exchange resins - properties and applications", 1975, VAN NOSTRAND REINHOLD COMPANY |
| S. AITIPAMULA ET AL.: "Polymorphs, Salts, and Cocrystals: What's in a Name?", CRYST. GROWTH DES., vol. 12, no. 5, 2012, pages 2147 - 2152, XP002733448, DOI: doi:10.1021/cg3002948 |
| SARAH J JACKSON: "The use of ion-exchange resins as potential bioadhesive drug delivery systems", UNIVERSITY OF NOTTINGHAM, 1999 |
| SERAJUDDIN ATM ET AL., PHARM RES, vol. 8, 1991, pages S103 |
| SOMCHAI SAWATDEE ET AL, SAUDI PHARMACEUTICAL JOURNAL., vol. 23, 2015, pages 504 - 514 |
| V RANADE; M A HOLLINGER: "Drug Delivery Systems", 2004, CRC PRESS |
| W HABIB ET AL., CRITICAL REVIEWS IN THERAPEUTIC DRUG CARRIER SYSTEMS, vol. 17, no. 1, 2000, pages 61 - 72 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019081451A1 (en) * | 2017-10-26 | 2019-05-02 | Mw Encap Limited | Liquid filled formulations of pde5 inhibitors |
| CN111356450A (en) * | 2017-10-26 | 2020-06-30 | 兆瓦恩卡普有限公司 | Liquid fill formulations of PDE5 inhibitors |
| JP2021054781A (en) * | 2019-09-26 | 2021-04-08 | デボン エルエス,リミテッド | Co-crystalline efinaconazole and method for producing the same |
| CN111388433A (en) * | 2020-03-27 | 2020-07-10 | 广州白云山医药集团股份有限公司白云山制药总厂 | Sildenafil citrate oral preparation and preparation method thereof |
| CN111388433B (en) * | 2020-03-27 | 2021-11-23 | 广州白云山医药集团股份有限公司白云山制药总厂 | Sildenafil citrate oral preparation and preparation method thereof |
| CN113694035A (en) * | 2021-10-09 | 2021-11-26 | 药大制药有限公司 | Preparation method of sildenafil tablets |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6314188B2 (en) | Tofacitinib oral sustained release dosage form | |
| KR101156916B1 (en) | Pharmaceutical compositions comprising imatinib and a release retardant | |
| JP4920798B2 (en) | Intraoral quick disintegrating tablet containing two or more kinds of particles | |
| KR20160045728A (en) | Pharmaceutical composition containing dimethyl fumarate for administration at a low daily dose | |
| WO2017168174A1 (en) | New pharmaceutical forms of sildenafil | |
| JP5479909B2 (en) | New formulation | |
| TW201304779A (en) | Pharmaceutical compositions | |
| JP2017533949A (en) | tablet | |
| TW201938564A (en) | Pharmaceutical composition including sodium alkyl sulfate | |
| ES2377552T3 (en) | Easily dissolved and stable compositions of candesartan cilexetil prepared with a wet granulation | |
| WO2014125352A1 (en) | Pharmaceutical compositions comprising tadalafil | |
| JP2020128442A (en) | Zinc acetate hydrate tablet and production method thereof | |
| WO2012020301A2 (en) | Oral controlled release pharmaceutical compositions of blonanserin | |
| JP2020169145A (en) | Pharmaceutical composition containing azilsartan | |
| JP2023071921A (en) | Lenalidomide oral tablet composition in various doses | |
| CA3085455C (en) | A solid oral dosage form comprising linagliptin | |
| WO2021106004A1 (en) | Pharmaceutical composition of s-adenosylmethionine | |
| JP6293850B1 (en) | Method for maintaining and stabilizing the rapid dissolution of azilsartan in a pharmaceutical composition | |
| JP2008546830A (en) | Sustained release formulation of active ingredient with pH-dependent solubility | |
| EP3154512A2 (en) | Solid oral formulations comprising solid melt dispersions of organic acids in xylitol | |
| EP2793856B1 (en) | Orally-disintegrating formulations of flurbiprofen | |
| WO2022034914A1 (en) | Readily-soluble solid pharmaceutical preparation and production method for same | |
| WO2022029798A1 (en) | Pharmaceutical compositions comprising ribociclib | |
| JP5321454B2 (en) | Manufacturing method of pharmaceutical tablets | |
| TW202408463A (en) | Pharmaceutical compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17716293 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17716293 Country of ref document: EP Kind code of ref document: A1 |