WO2010032128A1 - Pharmaceutical dosage forms comprising poly(e-caprolactone) - Google Patents
Pharmaceutical dosage forms comprising poly(e-caprolactone) Download PDFInfo
- Publication number
- WO2010032128A1 WO2010032128A1 PCT/IB2009/006917 IB2009006917W WO2010032128A1 WO 2010032128 A1 WO2010032128 A1 WO 2010032128A1 IB 2009006917 W IB2009006917 W IB 2009006917W WO 2010032128 A1 WO2010032128 A1 WO 2010032128A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dosage form
- extended release
- pharmaceutical dosage
- solid oral
- release pharmaceutical
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Definitions
- the present invention relates to pharmaceutical dosage forms, for example pharmaceutical dosage forms comprising poly( ⁇ -caprolactone), and processes of manufacture, uses, and methods of treatment thereof.
- Extended release oral dosage forms allow a specific release of active agent over an extended period of time. Larger dosing intervals, e.g. twice- or once-a- day dosing, may provide fewer side effects and overall better patient compliance.
- compositions and in particular extended release dosage forms which usually comprise a larger amount of active agent in a single dose are increasingly the subject of abuse.
- a particular dose of opioid agonist may be more potent when administered parenterally as compared to the same dose administered orally.
- Some formulations can be tampered with to provide the opioid agonist contained therein for illicit use.
- Controlled release opioid agonist formulations are sometimes milled or ground, and/or subject to extraction with solvents (e.g., ethanol) by drug abusers to provide the opioid contained therein for immediate release upon oral or parenteral administration.
- Extended release opioid agonist dosage forms which can liberate a portion of the opioid upon exposure to ethanol, can also result in a patient receiving the dose more rapidly than intended if a patient concomitantly uses alcohol with the dosage form.
- extended release pharmaceutical oral dosage forms There continues to exist a need in the art for extended release pharmaceutical oral dosage forms. In particular there continues to exist a need for such dosage forms that resist illicit use and are safe when concomitantly used with alcohol.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent, wherein at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of at least 10,000.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent, wherein poly( ⁇ -caprolactone) is present at an amount of at least about 50 weight-% of the extended release matrix formulation.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent, wherein the multi particulates have a diameter in the range of about 0.1 to about 3 mm.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent, and additionally comprising at least one high molecular weight polyethylene oxide.
- the invention encompasses a solid extended release pharmaceutical dosage form as described in the above paragraphs, wherein the active agent is an opioid analgesic, in particular selected from the group of codeine, morphine, oxycodone, hydrocodone, hydromorphone, or oxymorphone or pharmaceutically acceptable salts, hydrates and solvates thereof, and mixtures of any of the foregoing.
- the active agent is an opioid analgesic, in particular selected from the group of codeine, morphine, oxycodone, hydrocodone, hydromorphone, or oxymorphone or pharmaceutically acceptable salts, hydrates and solvates thereof, and mixtures of any of the foregoing.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent, wherein the dosage form provides release rates of the active agent in- vitro when measured by the USP Basket Method at 100 rpm at 900 ml simulated gastric fluid at 37 °C, between 12.5 % and 55 % (by wt) active agent released after 1 hour, between 25 % and 65 % (by wt) active agent released after 2 hours, between 45 % and 85 % (by wt) active agent released after 4 hours and between 55 % and 95 % (by wt) active agent released after 6 hours.
- These dosage forms comprise in particular oxycodone hydrochloride, hydromorphone hydrochloride, morphine sulfate or oxymorphone hydrochloride in the active agent.
- the invention encompasses a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent, wherein the dosage form provides release rates of the active agent in- vitro when measured by the USP Basket Method at 100 rpm at 900 ml simulated gastric fluid at 37 °C between 10 % and 30% (by wt) active agent released after 2 hour, 40 % and 75 % (by wt) active agent released after 8 hours and no less than 80%(by wt) active agent released after 22 hours.
- the invention further encompasses a method of treatment wherein a dosage form comprising an opioid analgesic as described herein is administered for treatment of pain to a patient in need thereof.
- the invention further encompasses the use of a dosage form comprising an opioid analgesic as described herein for the manufacture of a medicament for the treatment of pain.
- the invention further encompasses the use of poly( ⁇ -caprolactone) as matrix forming material in the manufacture of a solid extended release dosage form comprising an active agent selected from opioids for imparting to the solid extended release dosage form resistance to milling.
- the invention further encompasses a process of preparing a solid extended release pharmaceutical dosage form.
- the invention further encompasses a solid extended release pharmaceutical dosage form obtainable by a process as described herein.
- the solid extended release pharmaceutical dosage form is preferably an oral dosage form. According to certain embodiments of the invention the solid extended release pharmaceutical dosage form is for use as a suppository.
- extended release is defined for purposes of the present invention as to refer to products which are formulated to make the drug available over an extended period after ingestion thereby allowing a reduction in dosing frequency compared to a drug presented as a conventional dosage form (e.g. as a solution or an immediate release dosage form).
- immediate release is defined for the purposes of the present invention as to refer to products which are formulated to allow the drug to dissolve in the gastrointestinal contents without substantial delay or prolongation of the dissolution or absorption of the drug.
- solid oral extended release pharmaceutical dosage form for the purpose of the present invention refers to the administration form comprising a unit dose of active agent in extended release form such as an "extended release matrix formulation” and optionally other adjuvants and additives conventional in the art, such as a protective coating or a capsule and the like, and optionally any other additional features or components that are used in the dosage form.
- solid oral extended release pharmaceutical dosage form refers to said dosage form in intact form i.e. prior to any tampering.
- the extended release pharmaceutical dosage form can e.g. be a tablet comprising the extended release matrix formulation or a capsule comprising the extended release matrix formulation in the form of multi particulates.
- the "extended release pharmaceutical dosage form” may comprise a portion of active agent in extended release form and another portion of active agent in immediate release form, e.g. as an immediate release layer of active agent surrounding the dosage form or an immediate release component included within the dosage form.
- extended release matrix formulation is defined for purposes of the present invention as shaped solid form of a composition comprising at least one active agent and at least one extended release feature such as an extended release matrix material such as e.g. poly( ⁇ -caprolactone).
- the composition can optionally comprise more than these two compounds namely further active agents and additional retardants and/or other materials, including but not limited to high molecular weight polyethylene oxides and other adjuvants and additives conventional in the art.
- polyC ⁇ -caprolactone may for the purpose of the invention be abbreviated by PCL.
- the molecular weight of "poly( ⁇ -caprolactone)" for the purpose of the present invention relates to a number average molecular weight.
- Poly( ⁇ - caprolactone) is considered to have an approximate number average molecular weight of 10,000 when the viscosity is 400-1000 MPA at 25 degrees Celsius. Poly( ⁇ - caprolactone) is considered to have an approximate number average molecular weight of 37,000 when the melt flow index is 40g/10 minutes at 160 degrees Celsius and 2.16kg. Poly( ⁇ -caprolactone) is considered to have an approximate number average molecular weight of 42,500 when the melt flow index is 1.8 G/10 minutes at 80°C and 44 psi. Poly( ⁇ -caprolactone) is considered to have an approximate number average molecular weight of 80,000 when the melt flow index is 1.0 G/l 0 minutes at 80 degrees Celsius and 44 psi. .
- polyethylene oxide may for the purpose of the invention be abbreviated by PEO.
- PEO polyethylene oxide
- Compositions with lower molecular weight are usually referred to as polyethylene glycols.
- WO2008/023261 which is hereby incorporated by reference, describes pharmaceutical dosage forms prepared with PEO.
- high molecular weight polyethylene oxide is defined for proposes of the present invention as having an approximate molecular weight of at least 1,000,000.
- the approximate molecular weight is based on rheological measurements.
- Polyethylene oxide is considered to have an approximate molecular weight of 1,000,000 when a 2% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 1, at 10 rpm, at 25 0 C shows a viscosity range of 400 to 800 mPa s (cP).
- Polyethylene oxide is considered to have an approximate molecular weight of 2,000,000 when a 2% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 3, at 10 rpm, at 25°C shows a viscosity range of 2000 to 4000 mPa s (cP).
- Polyethylene oxide is considered to have an approximate molecular weight of 4,000,000 when a 1% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 2, at 2 rpm, at 25°C shows a viscosity range of 1650 to 5500 mPa s (cP).
- Polyethylene oxide is considered to have an approximate molecular weight of 5,000,000 when a 1% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 2, at 2 rpm, at 25°C shows a viscosity range of 5500 to 7500 mPa s (cP).
- Polyethylene oxide is considered to have an approximate molecular weight of 7,000,000 when a 1% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 2, at 2 rpm, at 25°C shows a viscosity range of 7500 to 10,000 mPa s (cP).
- Polyethylene oxide is considered to have an approximate molecular weight of 8,000,000 when a 1% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 2, at 2 rpm, at 25°C shows a viscosity range of 10,000 to 15,000 mPa s (cP).
- Polyethylene oxide is considered to have an approximate molecular weight of 100,000 when a 5% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVT, spindle No.
- polyethylene oxide is considered to have an approximate molecular weight of 900,000 when a 5% (by wt) aqueous solution of said polyethylene oxide using a Brookfield viscometer Model RVF, spindle No. 2, at 2 rpm, at 25°C shows a viscosity range of 8800 to 17,600 mPa s (cP).
- low molecular weight polyethylene oxide is defined for purposes of the present invention as having, based on the rheological measurements outlined above, an approximate molecular weight of less than 1,000,000.
- melt formed is defined for the purpose of the invention to relate to a process wherein an at least partially molten mass is formed and shaped. It includes without being limited to formed by extrusion, formed by casting and formed by injection molding.
- extrusion is defined for purposes of the present invention as referring to a process by which material is mixed and at least partially melted then forced through a die under controlled conditions.
- casting is defined for purposes of the present invention as referring to a process by which molten material is poured into a mold of a desired shape or onto a surface.
- injection molding is defined for purposed of the present invention as referring to a process by which molten material is injected under pressure into a mold.
- direct compression is defined for purposes of the present invention as referring to a tableting process wherein the tablet or any other compressed dosage form is made by a process comprising the steps of dry blending the compounds and compressing the dry blend to form the dosage form, e.g. by using a diffusion blend and/or convection mixing process (e.g. Guidance for Industry, SUPAC-IR/MR: Immediate Release and Modified Release Solid Oral Dosage Forms, Manufacturing Equipment Addendum).
- a diffusion blend and/or convection mixing process e.g. Guidance for Industry, SUPAC-IR/MR: Immediate Release and Modified Release Solid Oral Dosage Forms, Manufacturing Equipment Addendum.
- dosage forms are regarded as "resistant to milling" when the respective dosage form provides after milling an in- vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C, characterized by the percent amount of active released at 1 hour of dissolution that deviates no more than about 20 % points from the corresponding in- vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without milling.
- dosage forms are regarded as "resistant to grinding" when the respective dosage form provides after grinding an in-vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C, characterized by the percent amount of active released at 1 hour of dissolution that deviates no more than about 20 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without grinding.
- dosage forms are regarded as "resistant to alcohol extraction" when the respective dosage form provides an in-vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid comprising 40% ethanol at 37° C, characterized by the percent amount of active released at 1 hour of dissolution that deviates no more than about 20 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without ethanol.
- dosage forms are regarded as "resistant to milling and alcohol extraction" when the respective dosage form after milling provides an in-vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid comprising 40% ethanol at 37° C, characterized by the percent amount of active released at 1 hour of dissolution that deviates no more than about 20 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without ethanol and without milling.
- dosage forms are regarded as "resistant to grinding and alcohol extraction" when the respective dosage form after grinding provides an in-vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid comprising 40% ethanol at 37° C 5 characterized by the percent amount of active released at 1 hour of dissolution that deviates no more than about 20 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without ethanol and without grinding.
- SGF Simulated Gastric Fluid
- SLS sodium lauryl sulfate
- SGF sodium lauryl sulfate
- active agent is defined as a pharmaceutically active substance which includes without limitation opioid analgesics.
- opioid analgesic includes single compounds and combinations of compounds selected from the group of opioids and which provide an analgesic effect such as one single opioid agonist or a combination of opioid agonists, one single mixed opioid agonist-antagonist or a combination of mixed opioid agonist-antagonists, or one single partial opioid agonist or a combination of partial opioid agonists and combinations of an opioid agonists, mixed opioid agonist-antagonists and partial opioid agonists with one ore more opioid antagonists, stereoisomers, ether or ester, salts, hydrates and solvates thereof, compositions of any of the foregoing, and the like.
- the present invention disclosed herein is specifically meant to encompass the use of the opioid analgesic in form of any pharmaceutically acceptable salt thereof.
- Pharmaceutically acceptable salts include, but are not limited to, inorganic acid salts such as hydrochloride, hydrobromide, sulfate, phosphate and the like; organic acid salts such as formate, acetate, trifluoroacetate, maleate, tartrate and the like; sulfonates such as methanesulfonate, benzenesulfonate, p-toluenesulfonate, and the like; amino acid salts such as arginate, asparginate, glutamate and the like, and metal salts such as sodium salt, potassium salt, cesium salt and the like; alkaline earth metals such as calcium salt, magnesium salt and the like; organic amine salts such as triethylamine salt, pyridine salt, picoline salt, ethanolamine salt, triethanolamine salt, dicyclohexylamine salt, N,N'-dibenzylethylenediamine salt and the like.
- inorganic acid salts such as hydrochloride
- the opioids used according to the present invention may contain one or more asymmetric centers and may give rise to enantiomers, diastereomers, or other stereoisomeric forms.
- the present invention is also meant to encompass the use of all such possible forms as well as their racemic and resolved forms and compositions thereof.
- the compounds described herein contain olef ⁇ nic double bonds or other centers of geometric asymmetry, it is intended to include both E and Z geometric isomers. All tautomers are intended to be encompassed by the present invention as well.
- stereoisomers is a general term for all isomers of individual molecules that differ only in the orientation of their atoms is space. It includes enantiomers and isomers of compounds with more than one chiral center that are not mirror images of one another (diastereomers).
- chiral center refers to a carbon atom to which four different groups are attached.
- enantiomer or “enantiomeric” refers to a molecule that is nonsuperimposeable on its mirror image and hence optically active wherein the enantiomer rotates the plane of polarized light in one direction and its mirror image rotates the plane of polarized light in the opposite direction.
- racemic refers to a mixture of equal parts of enantiomers and which is optically inactive.
- resolution refers to the separation or concentration or depletion of one of the two enantiomeric forms of a molecule.
- Opioid agonists useful in the present invention include, but are not limited to, alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, buprenorphine, butorphanol, clonitazene, codeine, desomorphine, dextromoramide, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmorphine, etonitazene, etorphine, dihydroetorphine, fentanyl and derivatives, hydrocodone, hydromorphone, hydroxypethidine, isomethad
- Opioid antagonists useful in combination with opioid agonists as described above are e.g. naloxone, naltrexone and nalmephene or pharmaceutically acceptable salts, hydrates and solvates thereof, mixtures of any of the foregoing, and the like.
- a combination of oxycodone HCl and naloxone HCl in a ratio of about 2:1 is used.
- ratios of oxycodone HCl : naloxone HCl are 5: 2.5, 10:5, 20:10, 30:15, 40:20, 60:30, 80:40, 100:50 and 120:60.
- the opioid analgesic is selected from codeine, morphine, oxycodone, hydrocodone, hydromorphone, or oxymorphone or pharmaceutically acceptable salts, hydrates and solvates thereof, mixtures of any of the foregoing, and the like.
- the opioid analgesic is oxycodone, hydromorphone or oxymorphone or a salt thereof such as e.g. the hydrochloride.
- the dosage form comprises from about 5 mg to about 500 mg oxycodone hydrochloride, from about 1 mg to about 100 mg hydromorphone hydrochloride or from about 5 mg to about 500 mg oxymorphone hydrochloride. If other salts, derivatives or forms are used, equimolar amounts of any other pharmaceutically acceptable salt or derivative or form including but not limited to hydrates and solvates or the free base may be used.
- the dosage form may comprise e.g.
- the dosage form may comprise e.g. 5 mg, 7.5 mg, 10 mg, 15 mg, 20 mg, 30, mg, 40 mg, 45 mg, 50 mg, 60 mg, or 80 mg, 90 mg, 100 mg, 120 mg or 160 mg oxycodone hydrochloride or equimolar amounts of any other pharmaceutically acceptable salt, derivative or form including but not limited to hydrates and solvates or of the free base.
- the dosage form may comprise e.g. 5 mg, 7.5 mg, 10 mg, 15 mg, 20 mg, 30, mg, 40 mg, 45 mg, 50 mg, 60 mg, or 80 mg, 90 mg, 100 mg, 120 mg or 160 mg oxymorphone hydrochloride or equimolar amounts of any other pharmaceutically acceptable salt, derivative or form including but not limited to hydrates and solvates or of the free base.
- the dosage form may comprise e.g. 2 mg, 4 mg, 5 mg, 8 mg, 12 mg, 15 mg, 16 mg, 24 mg, 25 mg, 32 mg, 48 mg, 50 mg, 64 mg or 75 mg hydromorphone hydrochloride or equimolar amounts of any other pharmaceutically acceptable salt, derivative or form including but not limited to hydrates and solvates or of the free base.
- WO 2005/097801 Al, US 7,129,248 B2 and US 2006/0173029 Al describe a process for preparing oxycodone hydrochloride having a 14-hydroxycodeinone level of less than about 25 ppm, preferably of less than about 15 ppm, less than about 10 ppm, or less than about 5 ppm, more preferably of less than about 2 ppm, less than about 1 ppm, less than about 0.5 ppm or less than about 0.25 ppm.
- ppm means "parts per million”.
- ppm means parts per million of 14-hydroxycodeinone in a particular sample product.
- the 14-hydroxycodeinone level can be determined by any method known in the art, preferably by HPLC analysis using UV detection.
- oxycodone hydrochloride is used having a 14- hydroxycodeinone level of less than about 25 ppm, preferably of less than about 15 ppm, less than about 10 ppm, or less than about 5 ppm, more preferably of less than about 2 ppm, less than about 1 ppm, less than about 0.5 ppm or less than about 0.25 ppm.
- therapeutically active agents may be used in accordance with the present invention, either in combination with opioids or instead of opioids.
- therapeutically active agents include antihistamines (e.g., dimenhydrinate, diphenhydramine, chlorpheniramine and dexchlorpheniramine maleate), non -steroidal anti-inflammatory agents (e.g., naproxen, diclofenac, indomethacin, ibuprofen, sulindac, Cox-2 inhibitors) and acetaminophen, anti-emetics (e.g., metoclopramide, methylnaltrexone), anti- epileptics (e.g., phenytoin, meprobmate and nitrazepam), vasodilators (e.g., nifedipine, papaverine, diltiazem and nicardipine), anti-tussive agents and expectorants (e.g.
- anti-asthmatics e.g. theophylline
- antacids e.g. theophylline
- anti-spasmodics e.g. atropine, scopolamine
- antidiabetics e.g., insulin
- diuretics e.g., ethacrynic acid, bendrofluthiazide
- anti-hypotensives e.g., propranolol, clonidine
- antihypertensives e.g., clonidine, methyldopa
- bronchodilatiors e.g., albuterol
- steroids e.g., hydrocortisone, triamcinolone, prednisone
- antibiotics e.g., tetracycline
- antihemorrhoidals hypnotics, psychotropics, antidiarrheals, mucolytics, sedatives, decongestants (e.
- the invention is directed to the use of Cox-2 inhibitors as active agents, in combination with opioid analgesics or instead of opioid analgesics, for example the use of Cox-2 inhibitors such as meloxicam (4-hydroxy-2- methyl-iV-(5-methyl-2-thiazolyl)-2H-l,2-benzothiazine-3-carboxamide-l,l-dioxide), as disclosed in U.S. Serial No. 10/056,347 and 11/825,938, which are hereby incorporated by reference, nabumetone (4-(6-methoxy-2-naphthyl)-2-butanone), as disclosed in U.S. Serial No.
- the present invention is also directed to the dosage forms utilizing active agents such as for example, benzodiazepines, barbiturates or stimulants such as amphetamines. These may be combined with the respective antagonists.
- active agents such as for example, benzodiazepines, barbiturates or stimulants such as amphetamines.
- benzodiazepines refers to benzodiazepines and drugs that are derivatives of benzodiazepine that are able to depress the central nervous system.
- Benzodiazepines include, but are not limited to, alprazolam, bromazepam, chlordiazepoxide, clorazepate, diazepam, estazolam, flurazepam, halazepam, ketazolam, lorazepam, nitrazepam, oxazepam, prazepam, quazepam, temazepam, triazolam, methylphenidate as well as pharmaceutically acceptable salts, hydrates, and solvates and mixtures thereof.
- Benzodiazepine antagonists that can be used in the present invention include, but are not limited to, flumazenil as well as pharmaceutically acceptable salts, hydrates, and solvates.
- Barbiturates refer to sedative-hypnotic drugs derived from barbituric acid
- Barbiturates include, but are not limited to, amobarbital, aprobarbotal, butabarbital, butalbital, methohexital, mephobarbital, metharbital, pentobarbital, phenobarbital, secobarbital and as well as pharmaceutically acceptable salts, hydrates, and solvates mixtures thereof.
- Barbiturate antagonists that can be used in the present invention include, but are not limited to, amphetamines as well as pharmaceutically acceptable salts, hydrates, and solvates.
- Stimulants refer to drugs that stimulate the central nervous system.
- Stimulants include, but are not limited to, stimulants such as amphetamines, such as amphetamine, dextroamphetamine resin complex, dextroamphetamine, methamphetamine, methylphenidate as well as pharmaceutically acceptable salts, hydrates, and solvates and mixtures thereof.
- Stimulant antagonists that can be used in the present invention include, but are not limited to, benzodiazepines, as well as pharmaceutically acceptable salts, hydrates, and solvates as described herein.
- Fig. 1 to 14-1 depict the dissolution profiles of the respective Examples 1 to 14 as described below.
- Fig .14-2 depicts the intact (a), milled (b) and grinded(c) multiparticulates of Example 14
- Fig. 14-3 depicts the multiparticulates of Example 14 after milling in a coffee mill (a) and a comparison tablet after milling in a coffee mill (b).
- the invention relates to a solid extended release pharmaceutical dosage form, comprising a melt formed multi particulate extended release matrix formulation, comprising at least one poly( ⁇ - caprolactone), and at least one active agent.
- poly( ⁇ -caprolactone) is a suitable polymeric material for forming an extended release matrix formulation which can provide a wide variety of release profiles when used in the form of melt formed multi particulates.
- the melt forming according to the invention can be accomplished by several methods, including extrusion, casting and injection molding.
- the multi particulates have preferably a diameter in the range of about 0.1 to about 3 mm.
- poly( ⁇ -caprolactone) due to its specific polymer characteristics, imparts a milling and/or grinding resistance to the extended release formulation in that the multi particles comprising poly( ⁇ -caprolactone) do not form during milling and/or grinding smaller individual particles but in case of milling tend to fuse/melt together forming a lumpy mass and in case of grinding might deform.
- Figures 14-2 and 14-3 It is believed that the release of the active agent does therefore not substantially change upon milling or grinding. In some cases the release is even slowed down. Thereby the extended release dosage form comprising said multi particulates is rendered less attractive for abuse.
- At least one poly( ⁇ - caprolactone) with an approximate number average molecular weight of at least about 6,000 is used.
- the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of at least about 10,000.
- the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of at least about 20,000.
- the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of at least about 25,000.
- the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of at least about 37,000.
- the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of about 42,500. According to certain embodiments of the invention the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of at least about 80,000. According to further certain embodiments of the invention, the at least one poly( ⁇ -caprolactone) has an approximate number average molecular weight of between about 6,000 to about 80,000, or between about 10,000 and about 80,000, or between about 20,000 and about 80,000, or between about 25,000 and about 80,000or between about 37,000 and about 80,000, or between about 42,500 and about 80,000.
- the extended release matrix formulation comprises at least two poly( ⁇ -caprolactone) with an approximate number average molecular weight of between about 6,000 and about 25,000 and between about 37,000 and about 80,000, or between about 10,000 and about 25,000 and between about 37,000 and about 80,000, or between about 10,000 and about 25,000 and between about 42,500 and about 80,000.
- the overall content of poly( ⁇ -caprolactone) is at least about 50 weight-%, or at least about 60 weight-%, or at least about 70 weight-%, or at least about 80 weight-%, or at least about 90 weight-%, or between about 50 and about 90 weight-%, or between about 60 and about 90 weight-%, or between about 70 and about 90 weight-%, or between about 80 and about 90 weight-% of the extended release matrix formulation.
- the extended release matrix formulation comprises least one poly( ⁇ -caprolactone) with an approximate number average molecular weight of between about 37,000 and about 80,000 which is present at an amount of between about 50 and about 90 weight-% of the extended release matrix formulation.
- the extended release matrix formulation comprises further at least one polyethylene glycol, which may be present at an amount of between about 1 and about 20 or about 1 and about 15 weight-%.
- the extended release matrix formulation comprises further at least one high molecular weight polyethylene oxide with an approximate molecular weight of between about 1,000,000 and about 10,00O 5 OOO, based on rheological measurements. It is the finding of the inventors that the combination of poly( ⁇ -caprolactone) and high molecular polyethylene oxide provide a resistance to milling and/or grinding in combination with a resistance to alcohol extraction thereby rendering the dosage form less attractive for illicit use and rendering the dosage form safer when used in combination with alcohol.
- the high molecular weight polyethylene oxide may be present at an amount of between about 5 and about 35 weight-%.
- the high molecular weight polyethylene oxide used has been screened through a screen with a size of 15/100 of the average diameter of the resulting melt formed multi particulate extended release formulation. According to certain embodiments the high molecular weight polyethylene oxide used has been screened with a 100 US mesh screen.
- the extended release matrix formulation further comprises at least one poloxamer.
- the extended release matrix formulations may comprise further any other ingredients/excipients as conventional in the art.
- the active agent is an opioid analgesic, in particular selected from the group of alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, buprenorphine, butorphanol, clonitazene, codeine, desomorphine, dextromoramide, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmorphine, etonitazene, etorphine, dihydroetorphine, fentanyl and derivatives, hydrocodone, hydromorphone, hydroxypethidine
- the opioid analgesic is selected from the group of codeine, morphine, oxycodone, hydrocodone, hydromorphone, or oxymorphone or pharmaceutically acceptable salts, hydrates and solvates thereof, mixtures of any of the foregoing.
- the opioid analgesic is oxycodone hydrochloride and the dosage form comprises from about 5 mg to about 500 mg of oxycodone hydrochloride or in particular comprises 5 mg, 7.5 mg, 10 mg, 15 mg, 20 mg, 30, mg, 40 mg, 45 mg, 50 mg, 60 mg, or 80 mg, 90 mg, 100 mg, 120 mg or 160 mg of oxycodone hydrochloride.
- the oxycodone hydrochloride has a 14-hydroxycodeinone level of less than about 25 ppm, preferably of less than about 15 ppm, less than about 10 ppm, or less than about 5 ppm, or even less than 1 ppm.
- the opioid analgesic is oxymorphone hydrochloride and the dosage form comprises from about 1 mg to about 500 mg of oxymorphone hydrochloride, in particular 5 mg, 7.5 mg, 10 mg, 15 mg, 20 mg, 30 mg, 40 mg, 45 mg, 50mg, 60 mg, or 80 mg, 90 mg, 100 mg, 120 mg or 160 mg of oxymorphone hydrochloride.
- the opioid analgesic is hydromorphone hydrochloride and the dosage form comprises from about 1 mg to about 100 mg of hydromorphone hydrochloride, in particular 2 mg, 4 mg, 5 mg, 8 mg, 12 mg, 15 mg, 16 mg, 24 mg, 25 mg, 32 mg, 48 mg 50 mg, 64 mg or 75 mg of hydromorphone hydrochloride.
- the dosage form contains active in immediate release form, wherein the same or different active agents are in extended release and in immediate release form.
- the dosage form provides release rates of the active agent in- vitro when measured by the USP Basket Method at 100 rpm at 900 ml simulated gastric fluid at 37 °C, between about 12.5 % and about 55 % (by wt) active agent released after 1 hour, between about 25 % and about 65 % (by wt) active agent released after 2 hours, between about 45 % and about 85 % (by wt) active agent released after 4 hours and between about 55 % and about 95 % (by wt) active agent released after 6 hours.
- the dosage form provides release rates of the active agent in- vitro when measured by the USP Basket Method at 100 rpm at 900 ml simulated gastric fluid at 37 °C between about 10 % and about 30% (by wt) active agent released after 2 hour, about 40 % and about 75 % (by wt) active agent released after 8 hours and no less than about 80%(by wt) active agent released after 22 hours.
- the dosage form provides an in- vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid comprising 40% ethanol at 37° C, characterized by the percent amount of active agent released at 1 hour of dissolution that deviates no more than about 20 % points or no more than about 10 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without ethanol.
- the dosage form provides after milling an in- vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C, characterized by the percent amount of active agent released at 1 hour of dissolution that increases no more than about 20 % points or no more than 10 % points or even decreases when compared to the corresponding in- vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without milling.
- the dosage form provides after grinding an in-vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C, characterized by the percent amount of active agent released at 1 hour of dissolution that increases no more than about 20 % points or more than about 10 % points or even decreases when compared to the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid at 37° C without grinding.
- the dosage form after milling provides an in-vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid comprising 40% ethanol at 37° C, characterized by the percent amount of active agent released at 1 hour of dissolution that deviates no more than about 20 % points or no more than 10 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid without ethanol at 37° C without milling.
- the dosage form after grinding provides an in- vitro dissolution rate, when measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid, comprising 40% ethanol, at 37° C, characterized by the percent amount of active agent released at 1 hour of dissolution that deviates no more than about 20 % points or no more than 10 % points from the corresponding in-vitro dissolution rate measured in a USP Apparatus 1 (basket) at 100 rpm in 900 ml simulated gastric fluid without ethanol at 37° C without grinding.
- the active agent is oxycodone hydrochloride, hydromorphone hydrochloride or oxymorphone hydrochloride.
- the dosage forms as described herein are used in a method of treating pain in a patient in need thereof, wherein the dosage form comprises an opioid analgesic and the use of such a dosage form for the manufacture of a medicament for the treatment of pain.
- poly( ⁇ -caprolactone) is used as matrix forming material in the manufacture of a solid extended release oral dosage form comprising an active agent selected from opioids for imparting to the solid extended release oral dosage form resistance to milling and/or grinding.
- a process of preparing a solid oral extended release pharmaceutical dosage form comprising the steps of:
- a process of preparing a solid oral extended release pharmaceutical dosage form comprising the steps of:
- Extruding the screened and blended materials in a twin screw extruder fitted with a die and set on counter-rotation with zone (barrel) temperatures ranged from about 18°C to about 110°C to obtain strands with a
- the polyethylene oxide may be screened through a # 100 US mesh screen
- the solid oral extended release pharmaceutical dosage form is obtainable by a process as described above.
- composition of the poly( ⁇ -caprolactone) multiparticulates is summarized in Table 1 below.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C.
- the poly( ⁇ -caprolactone) pellets were difficult to grind and fused/melted during milling.
- composition of the poly( ⁇ -caprolactone) multiparticulates is summarized in Table 2 below.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C. 2. Sampling time - every minute up to 1440 minutes.
- Rotor Shaft Stainless steel 1.4571 Motor Speed, idle: 28000 revolutions/minute
- the poly( ⁇ -caprolactone) pellets were difficult to grind but did not fuse/melt during milling.
- composition of the poly( ⁇ -caprolactone) multi particulates is summarized in Table 3 below.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Thermodyne Hot Plate (temperature range 90° - 16O 0 C) until mixture appeared homogeneous.
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C. 2. Sampling time - every minute up to 1440 minutes.
- the poly( ⁇ -caprolactone) pellets were difficult to grind and fused/melted during milling.
- composition of the poly( ⁇ -caprolactone) multiparticulates is summarized in Table 4 below.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C.
- Chamber Volume 80 ml Blade: Stainless steel beater 1.4034
- the polyC ⁇ -caprolactone pellets were difficult to grind but did not fuse/melt during milling.
- composition of this poly( ⁇ -caprolactone) multiparticulate formulation is summarized in Table 5.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37 0 C.
- Milling Chamber Stainless steel
- the poly( ⁇ -caprolactone) pellets were tough. The pellets did not fuse/melt during milling. Table 5b
- composition of this poly( ⁇ -caprolactone) multiparticulate formulation is summarized in Table 6.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- PCL high moleculare weight polycaprolactne
- PCL high moleculare weight polycaprolactne
- BHT milled BHT
- Thermodyne Hot Plate (temperature range 90° - 16O 0 C) until mixture appeared homogeneous.
- Apparatus- USP Type I Baskets
- Sampling time every minute up to 1440 minutes.
- the poly( ⁇ -caprolactone) pellets were tough and difficult to grind. During milling the discrete matrix particles formed a single fused mass.
- composition of this poly( ⁇ -caprolactone) multiparticulate formulation is summarized in Table 7.
- Apparatus- USP Type I Baskets
- Chamber Volume 80 ml Blade: Stainless steel beater 1.4034
- composition of this poly( ⁇ -caprolactone) multiparticulate formulation is summarized in Table 8.
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C.
- SLS Sulfate
- EtOH Simulated Gastric Fluid with 40% ethanol
- Milling Chamber Stainless steel Chamber Volume: 80 ml
- the poly( ⁇ -caprolactone) pellets were difficult to grind and fused/melted during milling.
- composition of this poly( ⁇ -caprolactone) multiparticulate formulation is summarized in Table 9.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Thermodyne Hot Plate (temperature range 90° - 16O 0 C) until mixture appeared homogeneous.
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C. 2. Sampling time - every minute up to 1440 minutes.
- Blade Stainless steel beater 1.4034 Rotor Shaft: Stainless steel 1.4571 Motor Speed, idle: 28000 revolutions/minute Motor Speed, under load: 25000 revolutions/minute Circumferential Speed, idle: 76 m/s Circumferential Speed, under load: 53 m/s Motor rating input: 160 W Motor rating output: 100 W
- the poly( ⁇ -caprolactone) pellets were difficult to grind and fused/melted during milling.
- composition of this poly( ⁇ -caprolactone) multiparticulate formulation is summarized in Table 10.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37 0 C. 2. Sampling time - every minute up to 1440 minutes.
- Milling Chamber Stainless steel
- Motor rating input 160 W
- Motor rating output 100 W
- the poly( ⁇ -caprolactone) pellets were difficult to grind and fused/melted during milling.
- the dissolution results for milled samples are summarized in Figure 10a and Table 10b.
- the processing steps for manufacturing the poly( ⁇ -caprolactone) multiparticulates are as follows:
- PCL/PEO/BHT PCL/PEO/BHT and mixed on a Thermodyne Hot Plate (temperature range 90° - 16O 0 C) until the mixture appeared homogeneous. 5.
- Casting The molten drug/polymer blend was pressed between two stainless steel plates to a thickness of approximately 2 millimeters.
- Pelletizing The drug/polymer sheet was cut into pellets approximately 2 mm in length and width.
- Apparatus- USP Type I (Baskets), 100 rpm at 37 0 C. 2. Sampling time - every minute up to 1440 minutes.
- Rotor Shaft Stainless steel 1.4571 Motor Speed, idle: 28000 revolutions/minute
- composition of the poly( ⁇ -caprolactone) melt extruded multiparticulates is summarized in Table 12 below.
- Torque 97 Melt Pressure (psi): 1930 Feed rate (g/min.): 20 Screw speed (rpm): 66 Die Plate Hole diameter (mm): 1.0 (8-hole die plate)
- BHT were screened through a # 20 US mesh screen. 2. Blending: The materials screened in Step 1 were loaded into an 8 qt. V- blender QA Poly( ⁇ -caprolactone), Naltrexone HCl 5 Polyethylene oxide, BHT and 1 A Poly( ⁇ -caprolactone)) and blended for 10 minutes at ambient temperature.
- Extrusion Materials blended in Step 2 were metered into a twin screw extruder fitted with a die and processed into approximately 1 mm strands. The extruder was set on counter-rotation with zone (barrel) temperatures ranged from 18°C to 88°C.
- Cooling The strands were cooled on a conveyor at ambient temperature.
- Pelletizing The cooled strands were cut into pellets approximately 1 mm in length.
- Apparatus- USP Type I (Baskets), 100 rpm at 37 0 C.
- Chamber Volume 80 ml Blade: Stainless steel beater 1.4034
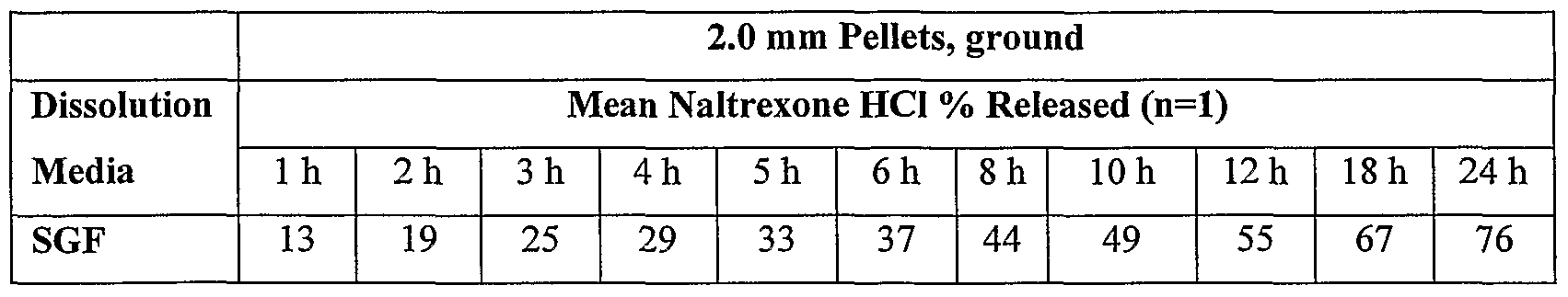
- the poly( ⁇ -caprolactone) pellets were difficult to crush with a mortar and pestle. They fused/melted during milling but incomplete after 15 seconds.
- composition of the Poly( ⁇ -caprolactone) melt extruded multiparticulates is summarized in Table 13 below.
- Torque (%): 62 Melt Pressure (psi) 370 Feed rate (g/min.): 22 Screw speed (rpm): 50 Melt Temp. ( 0 C): 89
- Blending The materials screened in Step 1 were loaded into an 8 qt. V-blender QA Poly( ⁇ -caprolactone), Naltrexone HCl, Polyethylene oxide, polyethylene glycol, BHT and 1 A Poly( ⁇ -caprolactone)) and blended for 10 minutes at ambient temperature.
- Extrusion Materials blended in Step 2 were metered into a twin screw extruder fitted with a die and processed into strands. The extruder was set on counter-rotation with zone (barrel) temperatures ranged from 18°C to 110°C.
- Cooling The strands were cooled on a conveyor at ambient temperature. 5.
- Pelletizing The cooled strands were cut into pellets approximately 1.0 mm, 1.5 mm and 2.0 mm in length for Sample #3, Sample #4 and Sample #1, respectively.
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C.
- Milling Chamber Stainless steel Chamber Volume: 80 ml Blade: Stainless steel beater 1.4034 Rotor Shaft: Stainless steel 1.4571 Motor Speed, idle: 28000 revolutions/minute
- Feed rate (g/min.): 20-22
- Blending The materials screened in Step 1 were loaded into a 16 qt. V-blender (/4 Poly( ⁇ -caprolactone), Oxycodone HCl, Polyethylene oxide, polyethylene glycol, BHT and 1 A Poly( ⁇ -caprolactone)) and blended for 10 minutes at ambient temperature.
- V-blender /4 Poly( ⁇ -caprolactone), Oxycodone HCl, Polyethylene oxide, polyethylene glycol, BHT and 1 A Poly( ⁇ -caprolactone)
- Extrusion Materials blended in Step 2 were metered into a twin screw extruder fitted with a die and processed into strands. The extruder was set on counter-rotation with zone (barrel) temperatures ranged from 14°C to 90°C. 4. Cooling: The strands were cooled on a conveyor at ambient temperature.
- Apparatus- USP Type I (Baskets), 100 rpm at 37 0 C. 2. Sampling time - every minute up to 1440 minutes.
- Extraction Solvent 1 :2 mixture acetonitrile and simulated gastric fluid without enzymes (SGF).
- Oxycodone N-oxide is the only known degradation product included in the % total degradation products.
- Noroxymorphone, oxymorphone, 10-hydroxyoxycodone, 6- ⁇ -oxycodol, 7,8-hydro-8, 14-dihydroxycodeinone, and hydrocodone which are known process impurities can be identified with this method but are not included in the calculation of % total degradation products.
- Extraction Solvent 1 :2 mixture acetonitrile and simulated gastric fluid without enzymes (SGF).
- Apparatus- USP Type I (Baskets), 100 rpm at 37°C. 2. Sampling Time- Generally, 1 hour, 2 hrs., 4 hrs., 8hrs., 12 hrs., 18 hrs. and 24 hrs.
Abstract
Description















































Claims
Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2737257A CA2737257C (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(.epsilon.-caprolactone) |
| KR1020117008610A KR101456741B1 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| JP2011527420A JP5539991B2 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage form comprising poly (ε-caprolactone) |
| US13/055,535 US20110165248A1 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| ES09786269.2T ES2586439T3 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly (E-caprolactone) |
| BRPI0913724A BRPI0913724B8 (en) | 2008-09-18 | 2009-09-17 | extended release dosage forms comprising naltrexone and poly (e-caprolactone) and their preparation |
| RU2011121010/15A RU2523896C2 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical drug forms, containing poly-(epsilon-caprolactone) |
| NZ592276A NZ592276A (en) | 2008-09-18 | 2009-09-17 | PHARMACEUTICAL DOSAGE FORMS COMPRISING POLY(epsilon-CAPROLACTONE) AND AN OPIOID |
| AU2009294308A AU2009294308B2 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| UAA201104686A UA101405C2 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| CN200980136742.4A CN102159193B (en) | 2008-09-18 | 2009-09-17 | Comprise the pharmaceutical dosage form of poly-(6-caprolactone) |
| EP09786269.2A EP2344136B1 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| MX2011002966A MX342643B (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone). |
| IL211314A IL211314A (en) | 2008-09-18 | 2011-02-20 | Pharmaceutical dosage forms comprising poly (epsilon-caprolactone) |
| ZA2011/01948A ZA201101948B (en) | 2008-09-18 | 2011-03-15 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9808908P | 2008-09-18 | 2008-09-18 | |
| US61/098,089 | 2008-09-18 | ||
| US22349709P | 2009-07-07 | 2009-07-07 | |
| US61/223,497 | 2009-07-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010032128A1 true WO2010032128A1 (en) | 2010-03-25 |
Family
ID=41435149
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/006917 WO2010032128A1 (en) | 2008-09-18 | 2009-09-17 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US20110165248A1 (en) |
| EP (1) | EP2344136B1 (en) |
| JP (1) | JP5539991B2 (en) |
| KR (1) | KR101456741B1 (en) |
| CN (1) | CN102159193B (en) |
| AU (1) | AU2009294308B2 (en) |
| BR (1) | BRPI0913724B8 (en) |
| CA (1) | CA2737257C (en) |
| CO (1) | CO6361903A2 (en) |
| ES (1) | ES2586439T3 (en) |
| IL (1) | IL211314A (en) |
| MX (1) | MX342643B (en) |
| NZ (2) | NZ592276A (en) |
| RU (1) | RU2523896C2 (en) |
| WO (1) | WO2010032128A1 (en) |
| ZA (1) | ZA201101948B (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8372432B2 (en) | 2008-03-11 | 2013-02-12 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| US8377453B2 (en) | 2008-03-11 | 2013-02-19 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| WO2013030267A1 (en) | 2011-08-30 | 2013-03-07 | Universiteit Gent | Multi-layered release formulation |
| WO2013084059A1 (en) * | 2011-12-09 | 2013-06-13 | Purdue Pharma L.P. | Pharmaceutical dosage forms comprising poly (epsilon- caprolactone) and polyethylene oxide |
| US8597681B2 (en) | 2009-12-22 | 2013-12-03 | Mallinckrodt Llc | Methods of producing stabilized solid dosage pharmaceutical compositions containing morphinans |
| WO2014006004A1 (en) | 2012-07-06 | 2014-01-09 | Egalet Ltd. | Abuse deterrent pharmaceutical compositions for controlled release |
| US8658631B1 (en) | 2011-05-17 | 2014-02-25 | Mallinckrodt Llc | Combination composition comprising oxycodone and acetaminophen for rapid onset and extended duration of analgesia |
| US8741885B1 (en) | 2011-05-17 | 2014-06-03 | Mallinckrodt Llc | Gastric retentive extended release pharmaceutical compositions |
| US8808745B2 (en) | 2001-09-21 | 2014-08-19 | Egalet Ltd. | Morphine polymer release system |
| US8821928B2 (en) | 2007-06-04 | 2014-09-02 | Egalet Ltd. | Controlled release pharmaceutical compositions for prolonged effect |
| US8858963B1 (en) | 2011-05-17 | 2014-10-14 | Mallinckrodt Llc | Tamper resistant composition comprising hydrocodone and acetaminophen for rapid onset and extended duration of analgesia |
| US8877241B2 (en) | 2003-03-26 | 2014-11-04 | Egalet Ltd. | Morphine controlled release system |
| US9005660B2 (en) | 2009-02-06 | 2015-04-14 | Egalet Ltd. | Immediate release composition resistant to abuse by intake of alcohol |
| US9023394B2 (en) | 2009-06-24 | 2015-05-05 | Egalet Ltd. | Formulations and methods for the controlled release of active drug substances |
| US9168228B2 (en) | 2009-02-06 | 2015-10-27 | Egalet Ltd. | Pharmaceutical compositions resistant to abuse |
| US9198861B2 (en) | 2009-12-22 | 2015-12-01 | Mallinckrodt Llc | Methods of producing stabilized solid dosage pharmaceutical compositions containing morphinans |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| EP3181124A1 (en) | 2015-12-16 | 2017-06-21 | Universität Basel | Abuse deterrent pharmaceutical dosage forms |
| US9694080B2 (en) | 2001-09-21 | 2017-07-04 | Egalet Ltd. | Polymer release system |
| US9700508B2 (en) | 2010-05-10 | 2017-07-11 | Euro-Celtique S.A. | Pharmaceutical compositions comprising hydromorphone and naloxone |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| US9814710B2 (en) | 2013-11-13 | 2017-11-14 | Euro-Celtique S.A. | Hydromorphone and naloxone for treatment of pain and opioid bowel dysfunction syndrome |
| US9901540B2 (en) | 2010-05-10 | 2018-02-27 | Euro-Celtique S.A. | Combination of active loaded granules with additional actives |
| US9993433B2 (en) | 2010-05-10 | 2018-06-12 | Euro-Celtique S.A. | Manufacturing of active-free granules and tablets comprising the same |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2611081C (en) * | 2005-06-03 | 2016-05-31 | Egalet A/S | A drug delivery system for delivering active substances dispersed in a dispersion medium |
| US8173666B2 (en) | 2007-03-12 | 2012-05-08 | Nektar Therapeutics | Oligomer-opioid agonist conjugates |
| US10512644B2 (en) | 2007-03-12 | 2019-12-24 | Inheris Pharmaceuticals, Inc. | Oligomer-opioid agonist conjugates |
| WO2010033195A1 (en) * | 2008-09-16 | 2010-03-25 | Nektar Therapeutics | Pegylated opioids with low potential for abuse |
| MX356111B (en) | 2012-04-18 | 2018-05-15 | SpecGx LLC | Immediate release, abuse deterrent pharmaceutical compositions. |
| BR112015021002B8 (en) | 2013-03-15 | 2023-03-28 | Mallinckrodt Llc | SOLID PHARMACEUTICAL DOSAGE FORM COMPRISING AN ACTIVE PHARMACEUTICAL INGREDIENT |
| US11617712B2 (en) | 2014-07-03 | 2023-04-04 | SpecGx LLC | Abuse deterrent immediate release formulations comprising non-cellulose polysaccharides |
| CN107405304B (en) * | 2015-02-04 | 2021-05-04 | 财团法人卫生研究院 | Sorbitan polyester complexes for stabilizing water-in-oil emulsions and controlled release of biologically active substances |
| MX2021002459A (en) | 2018-09-25 | 2021-04-29 | SpecGx LLC | Abuse deterrent immediate release capsule dosage forms. |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4148871A (en) * | 1977-10-11 | 1979-04-10 | Pitt Colin G | Sustained subdermal delivery ofdrugs using poly(ε-caprolactone) and its copolymers |
| US20050096388A1 (en) * | 1996-05-24 | 2005-05-05 | Angiotech International Ag | Compositions and methods for treating or preventing diseases of body passageways |
| WO2006082099A1 (en) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Break-resistant delayed-release forms of administration |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2557459B1 (en) * | 1984-01-02 | 1986-05-30 | Lhd Lab Hygiene Dietetique | POLYCAPROLACTONE-BASED INERT MATRIX FOR ORAL ADMINISTRATION OF A MEDICAMENT, AND METHOD FOR PREPARING THE GALENIC FORM COMPRISING THE SAME |
| US5716565A (en) * | 1993-09-27 | 1998-02-10 | Alfred University | Process for making ultra-fine stabilized zirconia particles |
| EP0804249A2 (en) * | 1994-03-15 | 1997-11-05 | Brown University Research Foundation | Polymeric gene delivery system |
| US5965161A (en) * | 1994-11-04 | 1999-10-12 | Euro-Celtique, S.A. | Extruded multi-particulates |
| ES2248908T7 (en) | 1997-06-06 | 2014-11-24 | Depomed, Inc. | Dosage forms of drugs orally and gastric retention for continued release of highly soluble drugs |
| RS49982B (en) * | 1997-09-17 | 2008-09-29 | Euro-Celtique S.A., | Synergistic analgesic combination of opioid analgesic and cyclooxygenase-2 inhibitor |
| UA73092C2 (en) * | 1998-07-17 | 2005-06-15 | Брістол-Майерс Сквібб Компані | Tablets with enteric coating and method for their manufacture |
| OA11728A (en) * | 1998-12-16 | 2005-02-01 | Aventis Pharma Inc | Biodegradable polymer encapsulated serotonin receptor antagonist and method for preparing the same. |
| EP2316440A1 (en) * | 2003-04-30 | 2011-05-04 | Purdue Pharma L.P. | Transdermal dosage form comprising an active agent component and an adverse agent component at the distal site of the active agent layer and one fluid communication between the surface of the active agent and the adverse agent |
| TWI357815B (en) * | 2003-06-27 | 2012-02-11 | Euro Celtique Sa | Multiparticulates |
| US20070048228A1 (en) * | 2003-08-06 | 2007-03-01 | Elisabeth Arkenau-Maric | Abuse-proofed dosage form |
| US20060194826A1 (en) * | 2003-09-25 | 2006-08-31 | Euro-Celtique S.A. | Pharmaceutical combinations of hydrocodone and naltrexone |
| TW201509943A (en) * | 2004-03-30 | 2015-03-16 | Euro Celtique Sa | Oxycodone hydrochloride composition, pharmaceutical dosage form, sustained release oral dosage form and pharmaceutically acceptable package having less than 25 PPM 14-hydroxycodeinone |
| US20080124400A1 (en) * | 2004-06-24 | 2008-05-29 | Angiotech International Ag | Microparticles With High Loadings Of A Bioactive Agent |
| SK286087B6 (en) * | 2004-08-18 | 2008-03-05 | Zentiva, A. S. | Method of preparation of oxycodone |
| CA2598204C (en) * | 2004-11-09 | 2015-01-13 | Board Of Regents, The University Of Texas System | Stabilized hme composition with small drug particles |
| CA2594373A1 (en) * | 2005-01-28 | 2006-08-03 | Euro-Celtique S.A. | Alcohol resistant dosage forms |
| WO2006125074A1 (en) * | 2005-05-17 | 2006-11-23 | Brown University Research Foundation | Drug delivery formulations for targeted delivery |
| RU2423997C9 (en) * | 2005-07-05 | 2011-11-20 | Эббетт ГмбХ унд Ко. КГ | Composition and dosage form containing solid or semisolid matrix |
| FR2901478B1 (en) * | 2006-05-24 | 2015-06-05 | Flamel Tech Sa | MULTIMICROPARTICULATED ORAL PHARMACEUTICAL FORM WITH PROLONGED RELEASE |
| SA07280459B1 (en) * | 2006-08-25 | 2011-07-20 | بيورديو فارما إل. بي. | Tamper Resistant Oral Pharmaceutical Dosage Forms Comprising an Opioid Analgesic |
| WO2008027442A2 (en) * | 2006-08-30 | 2008-03-06 | Theraquest Biosciences, Llc | Abuse deterrent oral pharmaceutical formulations of opioid agonists and method of use |
| NZ577560A (en) * | 2007-01-16 | 2012-01-12 | Egalet Ltd | Use of i) a polyglycol and ii) an active drug substance for the preparation of a pharmaceutical composition for i) mitigating the risk of alcohol induced dose dumping and/or ii) reducing the risk of drug abuse |
-
2009
- 2009-09-17 CA CA2737257A patent/CA2737257C/en active Active
- 2009-09-17 AU AU2009294308A patent/AU2009294308B2/en active Active
- 2009-09-17 NZ NZ592276A patent/NZ592276A/en not_active IP Right Cessation
- 2009-09-17 WO PCT/IB2009/006917 patent/WO2010032128A1/en active Application Filing
- 2009-09-17 ES ES09786269.2T patent/ES2586439T3/en active Active
- 2009-09-17 RU RU2011121010/15A patent/RU2523896C2/en not_active IP Right Cessation
- 2009-09-17 BR BRPI0913724A patent/BRPI0913724B8/en not_active IP Right Cessation
- 2009-09-17 NZ NZ601200A patent/NZ601200A/en not_active IP Right Cessation
- 2009-09-17 KR KR1020117008610A patent/KR101456741B1/en active IP Right Grant
- 2009-09-17 EP EP09786269.2A patent/EP2344136B1/en active Active
- 2009-09-17 US US13/055,535 patent/US20110165248A1/en not_active Abandoned
- 2009-09-17 JP JP2011527420A patent/JP5539991B2/en active Active
- 2009-09-17 MX MX2011002966A patent/MX342643B/en active IP Right Grant
- 2009-09-17 CN CN200980136742.4A patent/CN102159193B/en active Active
-
2011
- 2011-02-20 IL IL211314A patent/IL211314A/en active IP Right Grant
- 2011-03-15 ZA ZA2011/01948A patent/ZA201101948B/en unknown
- 2011-04-13 CO CO11045846A patent/CO6361903A2/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4148871A (en) * | 1977-10-11 | 1979-04-10 | Pitt Colin G | Sustained subdermal delivery ofdrugs using poly(ε-caprolactone) and its copolymers |
| US20050096388A1 (en) * | 1996-05-24 | 2005-05-05 | Angiotech International Ag | Compositions and methods for treating or preventing diseases of body passageways |
| WO2006082099A1 (en) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Break-resistant delayed-release forms of administration |
Non-Patent Citations (2)
| Title |
|---|
| BODMEIER R ET AL: "Preparation and characterization of microspheres containing the anti-inflammatory agents, indomethacin, ibuprofen, and ketoprofen", JOURNAL OF CONTROLLED RELEASE, ELSEVIER, AMSTERDAM, NL, vol. 10, no. 2, 1 November 1989 (1989-11-01), pages 167 - 175, XP025484215, ISSN: 0168-3659, [retrieved on 19891101] * |
| See also references of EP2344136A1 |
Cited By (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9694080B2 (en) | 2001-09-21 | 2017-07-04 | Egalet Ltd. | Polymer release system |
| US8808745B2 (en) | 2001-09-21 | 2014-08-19 | Egalet Ltd. | Morphine polymer release system |
| US9707179B2 (en) | 2001-09-21 | 2017-07-18 | Egalet Ltd. | Opioid polymer release system |
| US9884029B2 (en) | 2003-03-26 | 2018-02-06 | Egalet Ltd. | Morphine controlled release system |
| US9375428B2 (en) | 2003-03-26 | 2016-06-28 | Egalet Ltd. | Morphine controlled release system |
| US8877241B2 (en) | 2003-03-26 | 2014-11-04 | Egalet Ltd. | Morphine controlled release system |
| US9642809B2 (en) | 2007-06-04 | 2017-05-09 | Egalet Ltd. | Controlled release pharmaceutical compositions for prolonged effect |
| US8821928B2 (en) | 2007-06-04 | 2014-09-02 | Egalet Ltd. | Controlled release pharmaceutical compositions for prolonged effect |
| US8372432B2 (en) | 2008-03-11 | 2013-02-12 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| US8394408B2 (en) | 2008-03-11 | 2013-03-12 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| US8377453B2 (en) | 2008-03-11 | 2013-02-19 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| US8668929B2 (en) | 2008-03-11 | 2014-03-11 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| US9498446B2 (en) | 2009-02-06 | 2016-11-22 | Egalet Ltd. | Pharmaceutical compositions resistant to abuse |
| US9005660B2 (en) | 2009-02-06 | 2015-04-14 | Egalet Ltd. | Immediate release composition resistant to abuse by intake of alcohol |
| US10105321B2 (en) | 2009-02-06 | 2018-10-23 | Egalet Ltd. | Pharmaceutical compositions resistant to abuse |
| US9358295B2 (en) | 2009-02-06 | 2016-06-07 | Egalet Ltd. | Immediate release composition resistant to abuse by intake of alcohol |
| US9168228B2 (en) | 2009-02-06 | 2015-10-27 | Egalet Ltd. | Pharmaceutical compositions resistant to abuse |
| US9023394B2 (en) | 2009-06-24 | 2015-05-05 | Egalet Ltd. | Formulations and methods for the controlled release of active drug substances |
| US8597681B2 (en) | 2009-12-22 | 2013-12-03 | Mallinckrodt Llc | Methods of producing stabilized solid dosage pharmaceutical compositions containing morphinans |
| US9198861B2 (en) | 2009-12-22 | 2015-12-01 | Mallinckrodt Llc | Methods of producing stabilized solid dosage pharmaceutical compositions containing morphinans |
| US9700508B2 (en) | 2010-05-10 | 2017-07-11 | Euro-Celtique S.A. | Pharmaceutical compositions comprising hydromorphone and naloxone |
| US9901540B2 (en) | 2010-05-10 | 2018-02-27 | Euro-Celtique S.A. | Combination of active loaded granules with additional actives |
| US9993433B2 (en) | 2010-05-10 | 2018-06-12 | Euro-Celtique S.A. | Manufacturing of active-free granules and tablets comprising the same |
| US9433582B2 (en) | 2011-05-17 | 2016-09-06 | Mallinckrodt Llc | Gastric retentive extended release pharmaceutical compositions |
| US9539328B2 (en) | 2011-05-17 | 2017-01-10 | Mallinckrodt Llc | Tamper resistant composition comprising hydrocodone and acetaminophen for rapid onset and extended duration of analgesia |
| US9050335B1 (en) | 2011-05-17 | 2015-06-09 | Mallinckrodt Llc | Pharmaceutical compositions for extended release of oxycodone and acetaminophen resulting in a quick onset and prolonged period of analgesia |
| US8858963B1 (en) | 2011-05-17 | 2014-10-14 | Mallinckrodt Llc | Tamper resistant composition comprising hydrocodone and acetaminophen for rapid onset and extended duration of analgesia |
| US9468636B2 (en) | 2011-05-17 | 2016-10-18 | Mallinckrodt Llc | Combination composition comprising oxycodone and acetaminophen for rapid onset and extended duration of analgesia |
| US8741885B1 (en) | 2011-05-17 | 2014-06-03 | Mallinckrodt Llc | Gastric retentive extended release pharmaceutical compositions |
| US8658631B1 (en) | 2011-05-17 | 2014-02-25 | Mallinckrodt Llc | Combination composition comprising oxycodone and acetaminophen for rapid onset and extended duration of analgesia |
| US9629837B2 (en) | 2011-05-17 | 2017-04-25 | Mallinckrodt Llc | Pharmaceutical compositions for extended release of oxycodone and acetaminophen resulting in a quick onset and prolonged period of analgesia |
| JP2014525423A (en) * | 2011-08-30 | 2014-09-29 | ウニベルシテイト ヘント | Multi-layer release formulation |
| WO2013030267A1 (en) | 2011-08-30 | 2013-03-07 | Universiteit Gent | Multi-layered release formulation |
| AU2012327231B2 (en) * | 2011-12-09 | 2015-09-24 | Purdue Pharma L.P. | Pharmaceutical dosage forms comprising poly(epsilon-caprolactone) and polyethylene oxide |
| JP2015500273A (en) * | 2011-12-09 | 2015-01-05 | パーデュー、ファーマ、リミテッド、パートナーシップ | Pharmaceutical dosage forms comprising poly (epsilon-caprolactone) and polyethylene oxide |
| WO2013084059A1 (en) * | 2011-12-09 | 2013-06-13 | Purdue Pharma L.P. | Pharmaceutical dosage forms comprising poly (epsilon- caprolactone) and polyethylene oxide |
| US20200170954A1 (en) * | 2011-12-09 | 2020-06-04 | Purdue Pharma L.P. | Pharmaceutical dosage forms comprising poly(epsilon-caprolactone) and polyethylene oxide |
| US9549899B2 (en) | 2012-07-06 | 2017-01-24 | Egalet Ltd. | Abuse deterrent pharmaceutical compositions for controlled release |
| CN104684548A (en) * | 2012-07-06 | 2015-06-03 | 埃格勒特有限责任公司 | Abuse deterrent pharmaceutical compositions for controlled release |
| WO2014006004A1 (en) | 2012-07-06 | 2014-01-09 | Egalet Ltd. | Abuse deterrent pharmaceutical compositions for controlled release |
| US9044402B2 (en) | 2012-07-06 | 2015-06-02 | Egalet Ltd. | Abuse-deterrent pharmaceutical compositions for controlled release |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10639281B2 (en) | 2013-08-12 | 2020-05-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US9814710B2 (en) | 2013-11-13 | 2017-11-14 | Euro-Celtique S.A. | Hydromorphone and naloxone for treatment of pain and opioid bowel dysfunction syndrome |
| US10258616B2 (en) | 2013-11-13 | 2019-04-16 | Euro-Celtique S.A. | Hydromorphone and naloxone for treatment of pain and opioid bowel dysfunction syndrome |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10792254B2 (en) | 2013-12-17 | 2020-10-06 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| WO2017103111A1 (en) | 2015-12-16 | 2017-06-22 | Universität Basel | Abuse deterrent pharmaceutical dosage forms |
| EP3181124A1 (en) | 2015-12-16 | 2017-06-21 | Universität Basel | Abuse deterrent pharmaceutical dosage forms |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0913724B8 (en) | 2021-05-25 |
| CN102159193A (en) | 2011-08-17 |
| CO6361903A2 (en) | 2012-01-20 |
| KR20110055734A (en) | 2011-05-25 |
| JP2012502968A (en) | 2012-02-02 |
| RU2011121010A (en) | 2012-11-27 |
| KR101456741B1 (en) | 2014-10-31 |
| RU2523896C2 (en) | 2014-07-27 |
| MX2011002966A (en) | 2011-09-26 |
| ES2586439T3 (en) | 2016-10-14 |
| NZ592276A (en) | 2012-12-21 |
| ZA201101948B (en) | 2011-11-30 |
| US20110165248A1 (en) | 2011-07-07 |
| IL211314A0 (en) | 2011-04-28 |
| CN102159193B (en) | 2015-09-16 |
| AU2009294308B2 (en) | 2013-05-30 |
| BRPI0913724A2 (en) | 2017-06-20 |
| CA2737257A1 (en) | 2010-03-25 |
| EP2344136B1 (en) | 2016-06-15 |
| CA2737257C (en) | 2016-11-15 |
| IL211314A (en) | 2016-03-31 |
| AU2009294308A1 (en) | 2010-03-25 |
| MX342643B (en) | 2016-10-07 |
| EP2344136A1 (en) | 2011-07-20 |
| BRPI0913724B1 (en) | 2020-03-10 |
| JP5539991B2 (en) | 2014-07-02 |
| NZ601200A (en) | 2014-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2344136B1 (en) | Pharmaceutical dosage forms comprising poly(e-caprolactone) | |
| EP2787978B1 (en) | Pharmaceutical dosage forms comprising poly(epsilon-caprolactone) and polyethylene oxide | |
| EP2080514B1 (en) | Tamper resistant oral pharmaceutical dosage forms comprising an opioid analgesic | |
| JP5704789B2 (en) | Alcohol resistant dosage form | |
| AU2013203747B2 (en) | Pharmaceutical dosage forms comprising poly(e-caprolactone) | |
| AU2013200246B2 (en) | Tamper resistant oral pharmaceutical dosage forms comprising an opioid analgesic | |
| AU2017208264B2 (en) | Tamper resistant oral pharmaceutical dosage forms comprising an opioid analgesic | |
| JP2013100306A (en) | Alcohol resistance dosage form |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980136742.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09786269 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 211314 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2011527420 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2737257 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12011500547 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 0127011 Country of ref document: KE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/002966 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2121/DELNP/2011 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2011000223 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13105849 Country of ref document: CO Ref document number: 11045846 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 592276 Country of ref document: NZ Ref document number: 2009294308 Country of ref document: AU Ref document number: 2009786269 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20117008610 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2009294308 Country of ref document: AU Date of ref document: 20090917 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011121010 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0913724 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110318 |