WO2008140459A1 - Solid form - Google Patents
Solid form Download PDFInfo
- Publication number
- WO2008140459A1 WO2008140459A1 PCT/US2007/011707 US2007011707W WO2008140459A1 WO 2008140459 A1 WO2008140459 A1 WO 2008140459A1 US 2007011707 W US2007011707 W US 2007011707W WO 2008140459 A1 WO2008140459 A1 WO 2008140459A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- solid form
- fill material
- multiparticulate
- form according
- pressure sensitive
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4833—Encapsulating processes; Filling of capsules
Definitions
- the invention relates to a solid form comprising a film enrobing a compacted fill material, wherein the compacted fill material comprises a pressure sensitive multiparticulate and at least one cushioning agent and in which the multiparticulate and/or cushioning agent comprises an active material.
- the present invention is also directed to a method of making and using such a solid form.
- Active ingredients for example pharmaceutical, agrochemical and detergent active ingredients may be delivered through a wide range of solid forms including tablets and capsules.
- Conventional tablets generally are highly compacted and have relatively high densities.
- the active ingredient is generally compacted with other components in a blend to provide the requisite structural integrity for the tablet. Delivery of the active ingredient in use may however be unsatisfactory due to the compaction level and it is known to add excipients to the formulation to aid disintegration or dissolution of the tablet to improve delivery, aid compaction, increase strength and increase robustness of the solid form. This may however impose constraints on the flexibility of the formulator in developing tablets containing the active ingredient.
- Capsules generally include the active ingredient in a relatively non-compacted form.
- the lack of compaction together with the void space inherent within capsules mean that for a given large dose of active, the volume of the final solid form is greater than for more compacted solid forms. Increasing the size of the capsule to accommodate the required dose is undesirable for the user.
- capsules require a relatively high level of disintegrant to provide adequate disintegration of the solid form.
- Capsule shells may also be sensitive to moisture and present problems as regards storage and product shelf-life.
- WO 03/096963 discloses solid forms and processes utilizing films to enrobe a fill material to a degree of compaction less than that generally used to make a tablet.
- a solid form having a compacted fill material with a particular combination of components in which the components are less compacted than in a tablet but more than in a capsule formulation and which contains a pressure sensitive material in particulate form wherein each particle is composed of a plurality of smaller particles which are bound together, hereinafter referred to as a "multiparticulate", and a cushioning agent and in which the multiparticulate and/or cushioning agent comprises an active material ameliorate this problem.
- Sufficient pressure may ⁇ applied during the formation of the solid form to provide acceptable structural integrity without damaging the pressure sensitive component and adversely affecting the release profile of the active in the solid form.
- the invention provides in a first aspect a solid form comprising at least one film enrobing a compacted fill material wherein: i) the compacted fill material comprises a) a pressure sensitive multiparticulate; and b) at least one cushioning agent; ii) the pressure sensitive multiparticulate and/or the cushioning agent comprises at least one active material; iii) the solid form has a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number 1216; iv) the compacted fill material has a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa.
- multiparticulate has the meaning known to those killed in the art and refers to a material having discrete particles, each of which particle is itself composed of smaller particles which are bound together by physical or chemical interactions to produce the multiparticulate.
- multiparticulates include pellets, granules, spheres, microspheres, freeze dried material and crystals.
- the multiparticulate for use in the present invention may be coated or uncoated.
- Multiparticulates can have any shape and texture and can be produced by known processes. When taken orally, the multiparticulate suitably disperses freely in the gastrointestinal tract, optimizes absorption, and can minimize side effects.
- a multiparticulate may contain one or more components.
- pressure sensitive multiparticulate means a multiparticulate that has a physical attribute or characteristic for example its rate of dissolution, efficacy, or mechanical strength altered detrimentally to a material extent when the multiparticulate is compacted as compared to the uncompacted multiparticulate. Appropriate tests to determine whether an attribute or characteristic has been detrimentally affected as a result of compaction of the multiparticulate will depend on the particular characteristic being measured and are known to the skilled person.
- modified release refers to the solid form of the present invention having fast, pulsatile, delayed and/or controlled release characteristics as desired which have been modified as compared to an immediate release profile and are terms known in the art.
- immediate release solid form refers to a solid form in which the active material is released rapidly after administration.
- a typical release rate for an "immediate release” solid form is suitably not less than 85% active material release in 60 minutes, preferably in 45 minutes and especially in 30 minutes in the test specified in the USP Edition 29 Test Number -711 at page 2673 for said active material when said active material is placed in a dissolution medium as specified in the USP dissolution specification or selected from dissolution media specified in the USP according to the solubility properties of said active material. This is referred to in the USP as "Q" time.
- the term “immediate release” includes "fast release”.
- the solid form may comprise an active material which exhibits immediate release.
- the solid form may additionally comprise an active material which does not exhibit immediate release. If desired, the solid form may comprise an active material which exhibits immediate release and be free of an active material which does not exhibit immediate release.
- the at least one of the active material has a mean dissolution of at least 75% in 300 seconds in the test specified in the USP Edition 29 Test Number 711 at page 2673 for said active material when the active material is placed in a dissolution medium as specified in the USP dissolution specification or selected from dissolution media specified in the USP according to the solubility properties of the active material or as selected by the skilled person for example selected from: (i) the USP for the at least one active material, (U) water, (iii) 0.1 M HCI or (iv) phosphate buffer having a pH between 5.8 and 8.0.
- a solid form having an active material meeting this dissolution test is considered herein to be a "fast release" solid form.
- the solid form preferably comprises an active material exhibiting a fast release.
- the solid form may comprise a further active material which does not exhibit fast release.
- the solid form does not contain an active material which does not exhibit a fast release.
- Suitable active materials include a pharmaceutical active, food component or product, veterinary active, cosmetic component or product, an appetite suppressant, detergent component or product or nutraceutical component or product.
- the solid form comprises at least one film enrobing a compacted fill material wherein the compacted fill material comprises a pressure sensitive multiparticulate and at least one cushioning agent, the pressure sensitive multiparticulate and/or the cushioning agent comprising at least one active material, and the compacted fill material is selected from a pharmaceutical product, a food product, a veterinary product, a cosmetic, an appetite suppressant, a detergent product and a nutraceutical product, the said solid form shows a weight loss that is less than 1% during a 30 minutes USP friability test United States Pharmacopeia (USP) 29 Test Number 1216 (page 3046).
- USP United States Pharmacopeia
- the active material preferably is present as a component of the pressure sensitive multiparticulate or of the cushioning agent or both.
- the active material may perform the function of a pressure sensitive agent or a cushioning agent as desired and the multiparticulate or cushioning agent may consist of the active material.
- the film enrobing the compacted fill material is preferably a water-soluble film.
- the film is in intimate contact with the compacted fill material.
- intimate contact it is meant that the film and the compacted fill material are in direct contact preferably over the entire internal surface of film although some areas not being in direct contact with the compacted fill material may be acceptable.
- a tablet or other product form may be contained within the film or the film may have a lining or coating presenting a barrier between the compacted fill and the film.
- the invention also provides a method of making a solid form comprising at least one film enrobing a compacted fill material, the solid form having a weight loss that is less than 1% during a 30 minute friability test in accordance with United States
- the said method comprising: i) providing a first film shaped to define an interior volume for holding a compacted fill material and having an open end; ii) depositing a fill material in the interior volume the fill material comprising a pressure sensitive multiparticulate and a cushioning agent and wherein at least one of the said multiparticulate and the cushioning agent comprises at least one active material; iii) applying pressure to the fill material so as to compact the fill material; iv) applying a second film over the said open end to close the said open end; and v) sealing the first and second films together to enrobe the compacted fill material and provide the solid form.
- the invention provides a method of making a solid form comprising at least one film enrobing a compacted fill material, the solid form having a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number 1216 and the compacted fill material having a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa, the said method comprising: i) providing a first film shaped to define an interior volume for holding a compacted fill material and having an open end; ii) depositing via the open end a first zone of a first fill material in the interior volume; iii) depositing a second zone of a second fill material in the interior volume such that the interior volume comprises two zones of fill material wherein one of the first or second fill materials comprises a pressure sensitive multiparticulate and the other of the said first or second fill materials comprises a cushioning agent and wherein at
- the solid form of the present invention comprising a pressure sensitive multiparticulate can be obtained at very low compression forces using at least one cushioning agent. Despite these relatively low forces the resultant solid form retains low friability and high integral strength. In addition, it has been shown that such a solid form protects the pressure sensitive multiparticulate which is being enrobed. This is generally achieved by the combination of the low forces applied and the use of at least one cushioning agent.
- the solid form of the invention may be produced using low force arid at least one cushioning agent, that in combination, protect the pressure sensitive multiparticulate from damage so as to avoid or reduce changes to the manner in which the active material is released in use from the solid form due to the manufacturing process.
- the manner of release of the active material in use is referred to herein and in the art as the "release profile" of the active material..
- the compacted fill material preferably has a density of less than 1.1 g/ml and more preferably less than 1.05 g/ml.
- the density of the compacted fill material is suitably at least 0.55 g/ml, preferably, the density of the compacted fill material is from 0.55 to 1.04 g/ml, more preferably from 0.62 to 1.04 g/ml and desirably from 0.75 to 1 g/ml.
- the density of the solid form is suitably higher than that for conventional capsules and as the density contributes to the release profile of the solid form, this may be optimized by the formulator according to the release profile required.
- the compacted fill material suitably has a tensile strength of less than 0.9MPa, preferably less than 0.5MPa, especially less than 0.2MPa and particularly less than 0.1 MPa.
- the compacted fill material has sufficient tensile strength to retain the physical integrity of the compacted fill material and is preferably at least 0.05MPa.
- the robustness of the solid form is suitably provided by the enrobing film rather than by the compacted fill material.
- the solid form of the present invention has excellent robustness or physical strength.
- the robustness of a solid form may suitably be defined by measuring the weight loss of 10 solid forms when rotated in a USP friability apparatus. This test is as set out in USP 29 ⁇ 1216> p 3046.
- the solid form of the present invention shows a weight loss of less than 1% when tested for a 30 minutes in a friability drum.
- conventional solid forms such as coated tablets are considered to be robust when the weight loss after 4 minutes of friability testing is less than 1% measured according to USP 29 ⁇ 1216> p 3046, the solid form of the present invention is especially robust.
- the density of the compacted fill material of the solid form of the present invention refers to the total weight of the compacted fill material divided by the total volume of the solid form within the film material. This is typically referred to as the "apparent" density of the solid form. Unless otherwise stated or the context clearly requires, references to density herein are to "apparent" density.
- the apparent density of a conventional tablet is typically greater than 1 g/ml as disclosed in, Pharmaceutical Technology, 27 (4), 67-80.
- the fill material is lightly tamped so as to form a very weak slug that breaks up in the capsule shell, due to the air space above it.
- the density of the fill material is therefore similar to the bulk density of the loose powder.
- the latter is typically less than 0.5 g/ml as disclosed in, Pharmaceutical Technology, 27 (4), 67-80.
- the density of the compacted fill material of the present invention is at least 0.5 g/ml based on the total solid form volume.
- a typical method for determining the density D of the fill material in the present invention is to determine the fill weight W (1), the fill volume V, which depends on the size of the tooling used to manufacture the solid forms and to calculate D using equation (2) .
- W Wt-Wf (g), where Wt is the weight of the total enrobed solid form and Wf is the weight of the film enrobing the solid form.
- D W/V (g/ml)
- the volume V of the fill material is calculated using equation (3)
- V (212.7+110.8t)/1000 (ml), where t is the sidewall thickness of the solid form (mm), typically measured using a micrometer.
- the volume V of the fill material is calculated using equation (4):
- V [TT (13/2) 2 t]/1000 (ml), where t is the tablet thickness (mm), typically measured using a micrometer.
- Conventional tablets generally need to be robust for subsequent processing and handling such as film coating and packaging. Such tablets are considered to be robust when the tensile strength of the compacted material is at least 1.0 MPa for example as disclosed in Pharmaceutical Technology, p52-62, April 2005 (Douglas McCormick, - Evolutions in Direct Compression)..
- a typical method for determining the tensile strength for round flat faced cylinder shapes is to measure the crushing force (also called hardness) of compacts on a tablet hardness tester and calculate the tensile strength ⁇ using equation (5) for example as disclosed in Journal of Pharmaceutical Sciences, vol.
- ⁇ 2P / ⁇ Dt (MPa), where P is the crushing force (N), D is the compact diameter (mm), and t is the compact thickness (mm), typically measured using a micrometer.
- multiparticulate refers to a particulate material, each particle of which is composed of agglomerated smaller particles which are bound together by physical or chemical interactions to produce a particle having a plurality of smaller particles bound together.
- the multiparticulate may for example be in the form of pellets, micronised powders, granules, spheres, microspheres, freeze dried material or crystals.
- the multiparticulate may be coated or uncoated.
- a multiparticulate as employed in this invention may have any shape and texture and can be produced by a range of processes.
- a multiparticulate as employed in the present invention suitably disperses in the gastrointestinal tract and provides optimal absorption, and can minimize side effects.
- the multiparticulate may contain one or more components.
- At least one of said active material has a mean dissolution in the test specified in the USP Edition 29 Test Number 711 at page 2673 for said active material, which meets the USP dissolution specifications when said active material is placed in a dissolution medium as specified in the USP dissolution specification or selected from dissolution media specified in the USP according to the solubility properties of the active material.
- the at least one active material has a mean dissolution of at least 75% in 300 seconds when subjected to this dissolution test.
- dissolution medium is specified in the USP for an active material, this is suitably employed in the dissolution test. Where there is either: i) no USP test for the active material; ii) more than one test for the active material; or
- the active does not meet the USP specification with the specified medium; the skilled person will select the most appropriate medium for the dissolution test from the USP dissolution media specified in the USP having regard to the dissolution characteristics of the active material.
- media in which the dissolution test may be carried out include: (i) the medium specified in the USP preferably for said at least one active material, (ii) water, (iii) 0.1 M HCI or (iv) phosphate buffer having a pH between 5.8 and 8.0.
- An active which has a mean dissolution of at least 75% in 300 seconds when subjected to this dissolution test set out above is considered to provide a "fast release" profile.
- cushioning agent refers to a material, for example a cushion-like mass, capable of providing physical protection to the pressure sensitive multiparticulate by absorbing compaction stresses for example applied during manufacture or distribution such that attributes or characteristics of the multiparticulate are not adversely affected to a material extent.
- the solid form suitably contains at least two zones of compacted fill material.
- the zones may be of any shape or relative size and may have the same or different compositions but are suitably discrete.
- the zones are in the form of layers.
- a first layer comprises the cushioning agent and a second layer comprises the pressure sensitive multiparticulate.
- the solid form may have multiple layers of cushioning agent and multiple layers of pressure sensitive multiparticulate. The relative location of each layer in the solid form may be selected as appropriate to the application.
- a layer of pressure sensitive multiparticulate is disposed between two layers comprising a cushioning agent. Desirably, the multiparticulate is coated.
- the coated multiparticulate in combination with the low compaction level means that a disintegrant (to increase the disintegration rate of the solid form) need not be employed as part of the formulation as the multiparticulate fill, in use, disperses rapidly.
- the cushioning agent for example, when present in top and bottom layers of the compacted fill material and positioned next to the film or in a physical blend with the multiparticulate, suitably gives to the solid form added smoothness and appearance.
- the modified release solid form of the present invention contains a pressure sensitive multiparticulate in a physical blend with the cushioning agent.
- the blend of the multiparticulate and the cushioning agent is substantially homogeneous.
- the solid form may contain a mixture of the layered and blended embodiments of the invention as desired.
- the solid form may comprise at least one layer of the cushioning agent and a second layer comprising a physical bJend of pressure sensitive multiparticulate and cushioning agent.
- Suitable cushioning agents include, alone or in any combination acacia, alginic acid, calcium carbonate, calcium phosphate dibasic anhydrous, calcium phosphate dibasic dihydrate, calcium phosphate tribasic, carbomer, carboxymethylcellulose calcium, carboxymethylcellulose sodium, carrageenan, cellulose acetate, cellulose powdered, chitosan, citric acid, colloidal siiicon dioxide, croscarmelose sodium, crospovidone, dextrates, dextrine, dextrose, dicalcium phosphate, ethylcellulose, fructose, gelatin, glucose, glyceryl behenate, glyceryl palmitostearate, guar gum, hydroxyethyl cellulose, hydroxyethylmethyl cellulose, hydroxypropyl cellulose, hydroxypropylmethyl cellulose, kaolin, lactitol, lactose, lactose calcium carbonate, low substituted hydroxypropyl cellulose
- a layer of cushioning agent it is present in sufficient thickness to provide a cushioning effect during manufacture such that the release profile of the active is not materially altered.
- a first layer of cushioning agent has a thickness of at least 500 micrometers preferably at least 800 micrometers.
- second layer of cushioning agent suitably has at least one portion thereof having a thickness of at least 500 micrometers and preferably at least 800 micrometers.
- two or more cushioning layers are employed, they are suitably of approximately the same thickness although they may be of different thickness if desired.
- the pharmaceutical active can fall within any solubility class known in the art. It may include, for example, at least one pharmaceutical active that is a very soluble, freely soluble, soluble, poorly soluble or insoluble pharmaceutically active material.
- the active material has a solubility in water of 1 g in less than 1 g water, 1 g in 1 to 10 g water, 1 g active in 10 to 30 g water, 1 g active in 30 to 100 g water, 1 g active in 100 to 1,000 g water, 1 g active in 1 ,000 to 10,000 g water, and 1 g active in more than 10,000 g water.
- suitable classes of pharmaceutical actives include an analgesic, antiangina, antianaemia, antibiotic, antiarrhythmic, antidiarrheal, antidiuretic, antidepressant, antiemetic, antifungal, antirheumatic, antiviral, antiprotozoal, antihistamine, antihypertensive, anti-inflammatory, antimigraine, antinausea, antispasmodic, anxiolytic, beta blocker, calcium channel blocker, sedative, hypnotic, antipsychotic, bronchodilator, decongestant, cough expectorant, cough suppressant, antiasthma drug, corticosteroid, actives for treatment of cough or common cold, muscle relaxant, erectile dysfunction active, motion sickness active, anti-HIV, anti-malaria actives, anti-cholesterol actives, respiratory actives, gastronintestinal actives, cardiovascular actives, antidiabetes actives, central nervous system actives, anti- infection actives, mucolytics
- Suitable actives include paracetamol, pseudoephedrine, acravastine, lamivudine, abacavir, pravastatin, Roziglitazone, ezetimibe, Clavulanate, sulfamethoxazole, benazepril, Valsartan, Irbesartan, Losartan, Dutasteride, tamsolusin, Atazanavir, ritonavir, propoxyphene, Hydrocodone, Metocarbamol, Memantine, Donepezil, Glyburide, Pioglytazone, Glimepiride, Benazepril, Torcetrapib, Eprosartan, Telmisartan, Olmesartan, Lopinavir, Emtricitabine, Tenofovir, Amprenavir, Tipranavir, Atovaquone, Proguanil, 5-aminosalicylic acid, 4-aminophthalic acid, Bismuth citrate,
- the two or more actives may be from the same class or may be from different classes.
- combinations of active materials from different classes include an antibiotic in combination with one of a decongestant, an anti-inflammatory, a cough expectorant, a cough suppressant or an active for treatment of cough or common cold, a proton pump inhibitor, anti-hypertension and anti-cholesterol actives.
- classes where two or more active materials from one class may suitably be employed include respiratory actives, gastrointestinal actives, cardiovascular actives, antidiabetes actives, central nervous system actives, anti- infection actives, anti-viral actives, analgesics, anti-inflammatory actives, antibiotics, cough suppressants, expectorants, mucolytics, and nasal decongestants, anti-HIV , anti- malaria actives.
- Examples of particular combinations of active materials include: Paracetamol and Caffeine; Aspirin and paracetamol; Paracetamol and pseudoephedrine; Paracetamol and phenylephrine; lbuprofen and codeine; lbuprofen and pseudoephedrine; Paracetamol and diphenhydramine; Acravistine and pseudoephedrine; Paracetamol and dextromethorphan; Parcetamol and guaphenesin; Paracetamol, caffeine, aspirin; Aspirin and caffeine; Zidovudine, lamivudine and abacavir; Pravastatin and aspirin; Lamivudine and zidovudine; Roziglitazone and Metformin; Ezetimibe and fenofibrate; Amoxicillin and Clavulanate; Trimetoprim and sulfamethoxazole; Amlodipine and benazepril; Valsartan and Hydro
- the pressure sensitive multiparticulate may be coated if desired.
- Any conventional coating may be employed for example a sustained release coating, enteric coating, taste-masking coating, moisture barrier coating, pressure sensitive barrier coating, pressure insensitive barrier coating and oxygen barrier coating and combinations thereof.
- coating formulations suitably include at least one of methacrylates, ' methylcellulose, ethylcellulose, polyvinyl alcohol, hydroxypropylmethyl cellulose, hydroxypropyl cellulose, polyvinylacetate phthalate, methacrylic acid polymers, methacrylic ester copolymers, aminoalkyl methacrylate copolymers, hydroxypropylmethyl cellulose, carrageenan, ethylcellulose, starch acetates, polyethyl acrylate, polymethyl methacrylate, polymethacrylic acid, polyethyl acrylate, albumen, carboxymethyl cellulose, carboxymethylcellulose sodium, cellulose acetate, cellulose acetate phthalate, cetyl alcohol, chito
- the film to be used to enrobe the present invention may be any film capable of enrobing the compacted fill materials without adversely impacting the desired dissolution profile.
- the film to be used may comprise water soluble components, water insoluble components or may comprise soluble and insoluble components in combination.
- the film material may be designed to be fast, immediate, delayed, pulsatile or sustained release.
- the compacted fill material of the present invention is enrobed by a film comprising at least one water soluble polymer.
- Films generally useful in the present invention include those that are thermo formable and generally have a dissolution rate appropriate for the preparation of rapid release, preferably immediate release, solid forms of the invention.
- water soluble polymers include cellulosic materials such as hydroxyethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose; polyvinyl alcohol; hydrocolloids such as carrageenan, alginate and pectin; and water soluble acrylates.
- water insoluble polymers include ethylcellulose, methacrylates and cellulose acetate.
- the films used in the invention may be gelatin free.
- the films may contain plasticizers such as lactic acid, citric acid, polyethylene glycol, sorbitol, glycerine, triethylcitrate, propylene glycol, phthalates, triglycerides, triacetin, tributylcitrate, etc.
- plasticizers such as lactic acid, citric acid, polyethylene glycol, sorbitol, glycerine, triethylcitrate, propylene glycol, phthalates, triglycerides, triacetin, tributylcitrate, etc.
- WO 2004/026284, WO 02/083779 and WO 03/095548 disclose further examples of films that may be used in the invention and such are incorporated herein by reference. Examples of films that may be used in the present invention are available under the trade name XGEL UNO from BioTec Films LLC, Tampa, Florida, US. Films for use in the present invention may be made in a conventional manner. If desired, an
- solid forms of the present invention may be enrobed and prepared in accordance with WO 03/096963, WO 05/030115, WO 05/030116 and PCT/GB2005/001077 - all of which are incorporated herein by reference.
- the solid form of the present invention may be prepared using any equipment capable of enrobing a compacted fill material for use in the present invention
- the invention also provides for the use of a solid form according to the invention in which the active material comprises a pharmaceutical active in the manufacture of a medicament for treatment of the human or animal body by therapy.
- a further aspect of the invention provides a solid form according to the invention in which the active material comprises a pharmaceutical active for use in a method of treatment of the human or animal body by therapy.
- the active material comprises a pharmaceutical active for use in a method of treatment of the human or animal body by therapy.
- Figures 1 and 2 show microscope images of solid forms which have been coated.
- Figures 1 shows pellets according to the invention and
- Figure 2 shows damaged pellets presented for comparative purposes and are not according to the invention.
- Figures 3 to 5 show the release profile of a pharmaceutical active from a solid form according to the invention.
- the present invention is described by reference to the following illustrative examples. Unless otherwise indicated herein, all parts, percents, ratios and the like are by weight.
- Talc was homogenized in water using a homogenizer (Silverson L4, high shear mixer) for 10 minutes.
- the prepared suspension was added slowly into the Eudragit NE30D dispersion while stirring gently with a conventional low shear mixer. Finally the suspension was passed through a 0.5 mm sieve. The suspension was continuously stirred during the application of the coating.
- the LustreClear LC103 was dispersed in water using propeller mixer (IKA-WERKE, low shear mixer) for 50 minutes.
- Coating with the Eudragit NE30D suspension was performed under the following conditions: Inlet temperature: 26°C; Outlet temperature: 22°C; Spray rate: 3.0-4.7 g/min; Air across bed: 0.30 kPa; Atomization pressure: 1.5 bar; Spraying time: 85 minutes. Targeted weight gain was 12%.
- the coating with LustreClear LC103 was performed under the following conditions: Inlet temperature: 26°C; Outlet temperature: 25°C; Spray rate: 3.0 g/min; Air across bed: 0.35 kPa; Atomization pressure: 1.5 bar; Spraying time: 35 minutes; Drying time: 10 minutes in the fluid bed. Targeted weight gain was 1%.
- coatings with Eudragit NE30D and LustreClear LC103 were both done on the same pellets in the same coater, one after the other.
- LustreClear LC 103 is a second coating applied onto the Eudragit film. Its purpose is to decrease the sticking properties of the pellets coated with Eudragit NE30D.
- the talc was homogenized in water using a homogenizer (Silverson L4, high shear mixer) for 10 minutes.
- the prepared suspension was added slowly into the Eudragit RS/RL dispersion while stirring gently with a conventional stirrer. Finally the suspension was passed through a 0.5 mm sieve. The suspension was continuously stirred during the coating process.
- the LustreClear LC103 was dispersed in water using propeller mixer (IKA-WERKE, low shear mixer) for 50 minutes.
- the coating with Eudragit NE30D suspension was performed under the following conditions: Inlet temperature: 37°C; Outlet temperature: 27°C; Spray rate: 2.0-3.8 g/min; Air across bed: 0.30 kPa; Atomization pressure: 1.5 bar; Spraying time: ⁇ 180 minutes. Targeted weight gain was 10%.
- the coating with LustreClear LC103 was performed under the following conditions: Inlet temperature: 41 0 C; Outlet temperature: 29 0 C; Spray rate: 3.0 g/min; Air across bed: 0.35 kPa; Atomization pressure: 1.5 bar; Spraying time: ⁇ 35 minutes; Drying time: 10 minutes. Targeted weight gain was 1%.
- Fill material compacts manufacture Fill material compacts were prepared by compressing the fill material in an ESH compaction simulator (ESH Testing Ltd, Dudley, UK) with oblong flat punches having a dimension of 16.775 x 7.449 millimeters.
- the upper punch speed was 100 mm/sec and the cycle time was 0.36 second.
- the upper punch displacement was adjusted to vary the compaction force.
- the maximum applied pressure was chosen for the pellets- cushioning agent blend in order to obtain solid forms with good tensile strength. In the case of tabletting of the pellet/cushioning agent blend the pressure applied was around 177 MPa. Low compaction examples used for the compacted fill material was undertaken at a pressure of around 4.4 MPa.
- This low compaction pressure was used for the compression of: pellets without cushioning agent, pellets with cushioning agent (pellets - cushioning agent blend and pellets - cushioning agent - pellets layers).
- the weight of solid forms produced at low compaction pressure was varied according to the individual experiment. It was typically in the range between 350 and 800 milligrams.
- Six compacts were prepared for dissolution testing. The upper punch force and compact weight were recorded. The compaction pressure reported was based on the upper punch force divided by the punch area (113.03 square millimeters). Compacts were stored in double plastic bags prior to subsequent testing.
- Blending technique A measured amount of cushioning agent and coated pellets were loaded into the dye cavity. The blending was performed in the dye (manual blending).
- the hardness of the compacted dosages was evaluated on an Erweka hardness tester.
- Two-sample t-test is used to determine if two population means are equal. A common application of this is to test if a new process or treatment is superior to a current process or treatment. (Montgomery D.C. (2001), Design and Analysis of Experiments, John Wiley & Sons, INC, 35-36, 640)
- Soluble films known as XGEL UNO and supplied by Bio Tec Films LLC were cut into strips 6 centimeters by 20 centimeters approximately.
- the lower and upper films had a thickness of about 120 microns.
- the lower film was heated sufficiently to thermoform under vacuum into dose cups about 3 millimeters in height to conform to cavities (7.5 millimeters width by 16.75 length millimeters) with the cavity depth determined by height- adjustable dose-shaped lower pistons within the stainless steel die.
- the film strip was placed over the die and brought in contact with a heated TEFLON® coated surface by means of upward vacuum.
- the film was then drawn into the stainless steel die cavities by inverting the vacuum to form a strip of twelve thermoformed dose cups with 3.0 millimeters separation between adjacent dose cups.
- the fill composition was dosed by volume using a Kinematics and controls dosing system. Solid forms were manufactured using the layering technique with three different fill layers: 1) 100 mg layer of Avicel PH200; 2) 150 mg layer of Theophylline pellets coated with Eudragit NE30D; 3) 100 mg layer of Avicel PH200.
- the multiple doses were filled one on top of the others so as to form horizontal layers. Then the layered fill was lightly compacted in the dose cups with upper pistons, and the lower film was cut to separate the individual solid forms. The solid forms were then lifted by the lower pistons to expose a portion of the solid form sidewalls for application of the upper film to complete the enrobing of the solid form.
- An adhesive composed of 5% Methocel E15LV Premium, 45% Benzyl alcohol and 50% Triacetin, was applied (by transfer roller) to the upper filmstrip on the side to be pressed against the exterior sidewall of the dose cup. The upper film was placed over the solid forms containing the compressed layered powder fill and the film was heated by contact with the heating element using upper vacuum.
- the heated upper film was formed around the solid forms using the lower vacuum enclosing the fill material within the solid form by overlapping the upper film onto the sidewall of the solid forms.
- the top film was cut to separate the completed enrobed solid forms and the unused film was removed.
- the solid forms were further sealed by forcing them through a heated die under low pressure so that the cut film overlapping the sides was pressed smooth. All examples below used the apparatus set forth in WO 2005/030115,
- Soluble HPMC containing films were used to enrobe the solid forms.
- Dissolution was determined according to United States Pharmacopeia LISP 24 with dissolution apparatus 1 , baskets. The sampling was undertaken at the following intervals of time: time zero then at intervals of 30 min, 30 min, 1 hour, 2 hours and every 2 hours up to the end point of the given dissolution.
- the theophylline pressure sensitive multiparticulates were coated on the
- CoP coated pellets damaged pellets could not be evaluated
- Samples 1 and 6 in Table 6 did not contain any cushioning agent. Sample 1 did not undergo any compression and served as a control sample. During the compression of Samples 3-6 the compaction simulator settings were adjusted in accordance with the pressures set forth in Table 6. The compacts produced at the low compaction pressure (4.4 - 5.6 MPa) had a very low hardness / tensile strength and in order to withstand the stress applied to the compact during post compaction handling a supporting enrobing film would be needed. Compaction pressures of 4.4 MPa would be expected to have a solid form density of lower than 1.1 g/ml, while a compaction pressure of 176.9 MPa (Sample 5) would be expected to provide an apparent density of greater than 1.1 g/ml.
- the drug release profiles for different samples set forth in Table 6 were evaluated as follows in Table 7 and illustrated in Figure 3
- Sample 3 (150 mg pellets) 4.4 MPa mean 0.00 1 10.36 17.60 24.30 37.71 47.95 54.41 60.19 64.77 67.68 70.96 75.13 77.88 80.13 82.00
- Blend compaction 150 mg pellets
- MPa mean 0.00 11.15 17.95 25.16 36.41 45.47 54.60 60.40 65.51 69.69 73.99 75.29 76.92 78.25 79.97
- Blend compaction 150 mg pellets
- 176.9 MPa mean 0.00 11.19 25.74 40.55 65.52 80.79 87.04 92.05 95.89 96.78
- Coated pellets used in this Example 2 are from the same batch as the pellets coated in Example 1.
- Samples 1-3 in Table 14 are the same samples as Samples 1-3, respectively, in Table 6 of Example 1.
- the pellets in Sample 4 were compressed using the layering technique at the compaction pressure of 4.4 MPa and by varying the pellets layer quantity. These compacts consisted of a higher amount of coated pellets than the compacts in Samples 2-3. Table 14:
- CoP coated pellets
- Sample 1 did not undergo any compression and served as a control sample.
- Sample 2 (Table 14) was enrobed using the Enrobed solid form lab machine and Samples 3-4 (Table 14) were compressed using the compaction simulator.
- the cushioning agent was composed of two layers of Avicel PH200 (100 mg each). The amount of pellets in the layer varied from sample to sample.
- the compaction simulator settings were adjusted in order to obtain the compaction pressure of 4.4 MPa.
- Drug release profiles for different samples were evaluated using t- test analysis as set forth in Table 15. The data from Samples 1-3 in Table 14 is repeated from the same data in Table 7.
- Example 3 Coated pellets used in this Example 3 are from the same batch as the pellets coated in Example 1.
- the cushioning agent used was Avicel PH200. This example shows the effectiveness of different cushioning agents in the present invention. As the effect on release profile has been shown to be directly related to the degree of pellet coating damage, microscopic examination was undertaken (as it has previously been shown that this attribute is predictive of the drug release). Table 18
- Theophylline pellets (batch size of 1 kg), were coated with an Eudragit RS/ Eudragit RL polymer blend (formulation methods described above in Table 4). The pellets polymer weight gain was 6.6%. Top coating was LustreClear LC103 applied onto the pellets coated with Eudragit RS/ Eudragit RL. LustreClear weight gain was 1%. Total pellets weight gain after both coating was 11%w/w.
- Table 19 the ⁇ on compacted pellets (Sample 1) were compared to the pellets compacted with cushioning agent (Avicel PH200; Sample 2) using the layering technique and to the pellets compacted at 4.4 MPa without cushioning (Sample 3).
- CoP coated pellets
- CuA cushioning agent
- Sample 1 did not undergo any compression and served as a control sample.
- the compaction simulator settings were adjusted in order to obtain the desired compaction force.
- Table 20 Drug release profiles of evaluated samples time 0 0.5 1 2 4 6 8 10 12 14 16 (hours)
- a two-sample T-test was undertaken on Samples 1-3 to show the equivalency or non-equivalency, with statistical significance of the different drug release profile of Samples 2-3 evaluated versus the drug release profile of the non compacted pellets (Sample 1).
- the t test was performed on the results obtained from the dissolution at 4, 8 and 16 hours. In this case the first t-test was undertaken at the 4 hours time point because a significant lag time was observed in the release for the first 2 hours for all Samples 1-3 for this specific coating. Hence, a more significant data point is after the effects of this lag time have been taken into account.
Abstract
A solid form comprising at least one film enrobing a compacted fill material comprising a pressure sensitive multiparticulate and at least one cushioning agent, in which the multiparticulate and/or the cushioning agent comprises at least one active material, having low friability and wherein the compacted fill material has a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa.
Description
SOLID FORM
The invention relates to a solid form comprising a film enrobing a compacted fill material, wherein the compacted fill material comprises a pressure sensitive multiparticulate and at least one cushioning agent and in which the multiparticulate and/or cushioning agent comprises an active material. The present invention is also directed to a method of making and using such a solid form.
Active ingredients, for example pharmaceutical, agrochemical and detergent active ingredients may be delivered through a wide range of solid forms including tablets and capsules. Conventional tablets generally are highly compacted and have relatively high densities. In conventional tablets, the active ingredient is generally compacted with other components in a blend to provide the requisite structural integrity for the tablet. Delivery of the active ingredient in use may however be unsatisfactory due to the compaction level and it is known to add excipients to the formulation to aid disintegration or dissolution of the tablet to improve delivery, aid compaction, increase strength and increase robustness of the solid form. This may however impose constraints on the flexibility of the formulator in developing tablets containing the active ingredient.
Capsules generally include the active ingredient in a relatively non-compacted form. However, the lack of compaction together with the void space inherent within capsules mean that for a given large dose of active, the volume of the final solid form is greater than for more compacted solid forms. Increasing the size of the capsule to accommodate the required dose is undesirable for the user. Typically, capsules require a relatively high level of disintegrant to provide adequate disintegration of the solid form. Capsule shells may also be sensitive to moisture and present problems as regards storage and product shelf-life.
WO 03/096963 discloses solid forms and processes utilizing films to enrobe a fill material to a degree of compaction less than that generally used to make a tablet. It is specifically disclosed therein that because of the nature of the capsule produced that certain ancillary ingredients necessary in conventional tablet production may be omitted. It is further disclosed therein that, due to relatively loose compaction, components contained within a tablet which are "designed to disperse and breakup the tablet when it has reached the site of delivery, can be omitted, as the active ingredients in the capsule according to the present invention are in a non-compacted or at least less compacted form as compared to a conventional tablet, and this lesser compaction leads to the easy release and dispersal of active ingredients once the capsule film has dissolved, e.g., at the intended site of delivery".
There remains a need to provide a solid form able to deliver an active material, for example to provide a suitable dose level and a therapeutically beneficial delivery of the active material in a pharmaceutical product, and to retain flexibility for the formulator as to the components employed in the solid form. In making the solid form, application of sufficient pressure to the components in formation of the solid form to provide adequate structural integrity to the solid form may however be problematic due to the sensitivity to pressure of certain components. This sensitivity to pressure may result in the intended release profile of the active material being altered during production, distribution or storage prior to use.
The present inventors have found that a solid form having a compacted fill material with a particular combination of components in which the components are less compacted than in a tablet but more than in a capsule formulation and which contains a pressure sensitive material in particulate form wherein each particle is composed of a plurality of smaller particles which are bound together, hereinafter referred to as a "multiparticulate", and a cushioning agent and in which the multiparticulate and/or
cushioning agent comprises an active material, ameliorate this problem. Sufficient pressure may β applied during the formation of the solid form to provide acceptable structural integrity without damaging the pressure sensitive component and adversely affecting the release profile of the active in the solid form. The invention provides in a first aspect a solid form comprising at least one film enrobing a compacted fill material wherein: i) the compacted fill material comprises a) a pressure sensitive multiparticulate; and b) at least one cushioning agent; ii) the pressure sensitive multiparticulate and/or the cushioning agent comprises at least one active material; iii) the solid form has a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number 1216; iv) the compacted fill material has a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa.
The term "multiparticulate" is known to those skilled in the art. As used herein,
"multiparticulate has the meaning known to those killed in the art and refers to a material having discrete particles, each of which particle is itself composed of smaller particles which are bound together by physical or chemical interactions to produce the multiparticulate. Examples of multiparticulates include pellets, granules, spheres, microspheres, freeze dried material and crystals. The multiparticulate for use in the present invention may be coated or uncoated. Multiparticulates can have any shape and texture and can be produced by known processes. When taken orally, the multiparticulate suitably disperses freely in the gastrointestinal tract, optimizes
absorption, and can minimize side effects. A multiparticulate may contain one or more components.
As used herein, the term "pressure sensitive multiparticulate" means a multiparticulate that has a physical attribute or characteristic for example its rate of dissolution, efficacy, or mechanical strength altered detrimentally to a material extent when the multiparticulate is compacted as compared to the uncompacted multiparticulate. Appropriate tests to determine whether an attribute or characteristic has been detrimentally affected as a result of compaction of the multiparticulate will depend on the particular characteristic being measured and are known to the skilled person.
As used herein, the term "modified release" refers to the solid form of the present invention having fast, pulsatile, delayed and/or controlled release characteristics as desired which have been modified as compared to an immediate release profile and are terms known in the art. The term "immediate release" solid form as used herein refers to a solid form in which the active material is released rapidly after administration. A typical release rate for an "immediate release" solid form is suitably not less than 85% active material release in 60 minutes, preferably in 45 minutes and especially in 30 minutes in the test specified in the USP Edition 29 Test Number -711 at page 2673 for said active material when said active material is placed in a dissolution medium as specified in the USP dissolution specification or selected from dissolution media specified in the USP according to the solubility properties of said active material. This is referred to in the USP as "Q" time. The term "immediate release" includes "fast release".
The solid form may comprise an active material which exhibits immediate release. The solid form may additionally comprise an active material which does not
exhibit immediate release. If desired, the solid form may comprise an active material which exhibits immediate release and be free of an active material which does not exhibit immediate release.
Preferably, the at least one of the active material has a mean dissolution of at least 75% in 300 seconds in the test specified in the USP Edition 29 Test Number 711 at page 2673 for said active material when the active material is placed in a dissolution medium as specified in the USP dissolution specification or selected from dissolution media specified in the USP according to the solubility properties of the active material or as selected by the skilled person for example selected from: (i) the USP for the at least one active material, (U) water, (iii) 0.1 M HCI or (iv) phosphate buffer having a pH between 5.8 and 8.0.
A solid form having an active material meeting this dissolution test is considered herein to be a "fast release" solid form. The solid form preferably comprises an active material exhibiting a fast release. The solid form may comprise a further active material which does not exhibit fast release. As desired, the solid form does not contain an active material which does not exhibit a fast release.
Suitable active materials include a pharmaceutical active, food component or product, veterinary active, cosmetic component or product, an appetite suppressant, detergent component or product or nutraceutical component or product. Preferably, the solid form comprises at least one film enrobing a compacted fill material wherein the compacted fill material comprises a pressure sensitive multiparticulate and at least one cushioning agent, the pressure sensitive multiparticulate and/or the cushioning agent comprising at least one active material, and the compacted fill material is selected from a pharmaceutical product, a food product, a veterinary product, a cosmetic, an appetite suppressant, a detergent product and a nutraceutical product, the said solid form shows
a weight loss that is less than 1% during a 30 minutes USP friability test United States Pharmacopeia (USP) 29 Test Number 1216 (page 3046).
The active material preferably is present as a component of the pressure sensitive multiparticulate or of the cushioning agent or both. The active material may perform the function of a pressure sensitive agent or a cushioning agent as desired and the multiparticulate or cushioning agent may consist of the active material.
The film enrobing the compacted fill material is preferably a water-soluble film. Desirably, the film is in intimate contact with the compacted fill material. By "intimate contact" it is meant that the film and the compacted fill material are in direct contact preferably over the entire internal surface of film although some areas not being in direct contact with the compacted fill material may be acceptable. For example, a tablet or other product form may be contained within the film or the film may have a lining or coating presenting a barrier between the compacted fill and the film.
The invention also provides a method of making a solid form comprising at least one film enrobing a compacted fill material, the solid form having a weight loss that is less than 1% during a 30 minute friability test in accordance with United States
Pharmacopeia 29 Test Number 1216 and the compacted fill material having a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa, the said method comprising: i) providing a first film shaped to define an interior volume for holding a compacted fill material and having an open end; ii) depositing a fill material in the interior volume the fill material comprising a pressure sensitive multiparticulate and a cushioning agent and wherein at least one of the said multiparticulate and the cushioning agent comprises at least one active material; iii) applying pressure to the fill material so as to compact the fill material;
iv) applying a second film over the said open end to close the said open end; and v) sealing the first and second films together to enrobe the compacted fill material and provide the solid form.
In a further aspect, the invention provides a method of making a solid form comprising at least one film enrobing a compacted fill material, the solid form having a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number 1216 and the compacted fill material having a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa, the said method comprising: i) providing a first film shaped to define an interior volume for holding a compacted fill material and having an open end; ii) depositing via the open end a first zone of a first fill material in the interior volume; iii) depositing a second zone of a second fill material in the interior volume such that the interior volume comprises two zones of fill material wherein one of the first or second fill materials comprises a pressure sensitive multiparticulate and the other of the said first or second fill materials comprises a cushioning agent and wherein at least one of the said multiparticulate and the cushioning agent comprises at least one active material; iv) applying pressure to the fill material so as to compact the at least two zones of fill material v) applying a second film over the said open end to close the said open end; and
vii) sealing the first and second films together to enrobe the compacted fill material and provide the solid form.
Applicants have advantageously found that the solid form of the present invention comprising a pressure sensitive multiparticulate can be obtained at very low compression forces using at least one cushioning agent. Despite these relatively low forces the resultant solid form retains low friability and high integral strength. In addition, it has been shown that such a solid form protects the pressure sensitive multiparticulate which is being enrobed. This is generally achieved by the combination of the low forces applied and the use of at least one cushioning agent.
Advantageously, the solid form of the invention may be produced using low force arid at least one cushioning agent, that in combination, protect the pressure sensitive multiparticulate from damage so as to avoid or reduce changes to the manner in which the active material is released in use from the solid form due to the manufacturing process. The manner of release of the active material in use is referred to herein and in the art as the "release profile" of the active material..
The compacted fill material preferably has a density of less than 1.1 g/ml and more preferably less than 1.05 g/ml. The density of the compacted fill material is suitably at least 0.55 g/ml, preferably, the density of the compacted fill material is from 0.55 to 1.04 g/ml, more preferably from 0.62 to 1.04 g/ml and desirably from 0.75 to 1 g/ml. The density of the solid form is suitably higher than that for conventional capsules and as the density contributes to the release profile of the solid form, this may be optimized by the formulator according to the release profile required.
The compacted fill material suitably has a tensile strength of less than 0.9MPa, preferably less than 0.5MPa, especially less than 0.2MPa and particularly less than 0.1 MPa. The compacted fill material has sufficient tensile strength to retain the physical
integrity of the compacted fill material and is preferably at least 0.05MPa. . The robustness of the solid form is suitably provided by the enrobing film rather than by the compacted fill material.
The solid form of the present invention has excellent robustness or physical strength. The robustness of a solid form may suitably be defined by measuring the weight loss of 10 solid forms when rotated in a USP friability apparatus. This test is as set out in USP 29 <1216> p 3046. The solid form of the present invention shows a weight loss of less than 1% when tested for a 30 minutes in a friability drum. As conventional solid forms such as coated tablets are considered to be robust when the weight loss after 4 minutes of friability testing is less than 1% measured according to USP 29 <1216> p 3046, the solid form of the present invention is especially robust.
The density of the compacted fill material of the solid form of the present invention refers to the total weight of the compacted fill material divided by the total volume of the solid form within the film material. This is typically referred to as the "apparent" density of the solid form. Unless otherwise stated or the context clearly requires, references to density herein are to "apparent" density.
The apparent density of a conventional tablet is typically greater than 1 g/ml as disclosed in, Pharmaceutical Technology, 27 (4), 67-80. In a conventional hard capsule, the fill material is lightly tamped so as to form a very weak slug that breaks up in the capsule shell, due to the air space above it. In a conventional hard capsule, the density of the fill material is therefore similar to the bulk density of the loose powder. The latter is typically less than 0.5 g/ml as disclosed in, Pharmaceutical Technology, 27 (4), 67-80. The density of the compacted fill material of the present invention is at least 0.5 g/ml based on the total solid form volume. A typical method for determining the density D of the fill material in the present invention is to determine the fill weight W (1), the fill volume V, which depends on the
size of the tooling used to manufacture the solid forms and to calculate D using equation (2) .
(1) W = Wt-Wf (g), where Wt is the weight of the total enrobed solid form and Wf is the weight of the film enrobing the solid form. (2) D = W/V (g/ml)
For a solid form of the present invention having a 70 microns thick film and made with oblong concave tooling of 16.6 mm length and 7.3 mm width, the volume V of the fill material is calculated using equation (3)
(3) V = (212.7+110.8t)/1000 (ml), where t is the sidewall thickness of the solid form (mm), typically measured using a micrometer.
For a tablet or compact that is made using 13 mm diameter flat round punches, the volume V of the fill material is calculated using equation (4):
(4) V= [TT (13/2)2 t]/1000 (ml), where t is the tablet thickness (mm), typically measured using a micrometer. Conventional tablets generally need to be robust for subsequent processing and handling such as film coating and packaging. Such tablets are considered to be robust when the tensile strength of the compacted material is at least 1.0 MPa for example as disclosed in Pharmaceutical Technology, p52-62, April 2005 (Douglas McCormick, - Evolutions in Direct Compression).. A typical method for determining the tensile strength for round flat faced cylinder shapes is to measure the crushing force (also called hardness) of compacts on a tablet hardness tester and calculate the tensile strength σ using equation (5) for example as disclosed in Journal of Pharmaceutical Sciences, vol. 59 (5 ), 688-691 "Determination of tablet strength by the diametral-compression test", (Fell J. T. and Newton J. M., 1970), (5) σ = 2P / πDt (MPa), where P is the crushing force (N), D is the compact diameter (mm), and t is the compact thickness (mm), typically measured using a micrometer.
As used herein, the term "multiparticulate" refers to a particulate material, each particle of which is composed of agglomerated smaller particles which are bound together by physical or chemical interactions to produce a particle having a plurality of smaller particles bound together. The multiparticulate may for example be in the form of pellets, micronised powders, granules, spheres, microspheres, freeze dried material or crystals. The multiparticulate may be coated or uncoated. A multiparticulate as employed in this invention may have any shape and texture and can be produced by a range of processes.
When taken orally, a multiparticulate as employed in the present invention suitably disperses in the gastrointestinal tract and provides optimal absorption, and can minimize side effects. The multiparticulate may contain one or more components.
Suitably, at least one of said active material has a mean dissolution in the test specified in the USP Edition 29 Test Number 711 at page 2673 for said active material, which meets the USP dissolution specifications when said active material is placed in a dissolution medium as specified in the USP dissolution specification or selected from dissolution media specified in the USP according to the solubility properties of the active material. In an especially preferred embodiment, the at least one active material has a mean dissolution of at least 75% in 300 seconds when subjected to this dissolution test.
Where a dissolution medium is specified in the USP for an active material, this is suitably employed in the dissolution test. Where there is either: i) no USP test for the active material; ii) more than one test for the active material; or
Hi) the active does not meet the USP specification with the specified medium; the skilled person will select the most appropriate medium for the dissolution test from the USP dissolution media specified in the USP having regard to the dissolution characteristics of the active material.
Examples of media in which the dissolution test may be carried out include: (i) the medium specified in the USP preferably for said at least one active material, (ii) water, (iii) 0.1 M HCI or (iv) phosphate buffer having a pH between 5.8 and 8.0.
An active which has a mean dissolution of at least 75% in 300 seconds when subjected to this dissolution test set out above is considered to provide a "fast release" profile.
The term "cushioning agent" refers to a material, for example a cushion-like mass, capable of providing physical protection to the pressure sensitive multiparticulate by absorbing compaction stresses for example applied during manufacture or distribution such that attributes or characteristics of the multiparticulate are not adversely affected to a material extent.
In a preferred embodiment of the present invention, the solid form suitably contains at least two zones of compacted fill material. The zones may be of any shape or relative size and may have the same or different compositions but are suitably discrete. In a preferred embodiment, the zones are in the form of layers. Preferably, a first layer comprises the cushioning agent and a second layer comprises the pressure sensitive multiparticulate. The solid form may have multiple layers of cushioning agent and multiple layers of pressure sensitive multiparticulate. The relative location of each layer in the solid form may be selected as appropriate to the application. In an especially preferred embodiment, a layer of pressure sensitive multiparticulate is disposed between two layers comprising a cushioning agent. Desirably, the multiparticulate is coated. Advantageously, the coated multiparticulate in combination with the low compaction level means that a disintegrant (to increase the disintegration rate of the solid form) need not be employed as part of the formulation as the multiparticulate fill, in use, disperses rapidly.
Another added benefit of the present invention is that the cushioning agent, for example, when present in top and bottom layers of the compacted fill material and positioned next to the film or in a physical blend with the multiparticulate, suitably gives to the solid form added smoothness and appearance. In another embodiment of the present invention, the modified release solid form of the present invention contains a pressure sensitive multiparticulate in a physical blend with the cushioning agent. Preferably, the blend of the multiparticulate and the cushioning agent is substantially homogeneous.
The solid form may contain a mixture of the layered and blended embodiments of the invention as desired. For example, the solid form may comprise at least one layer of the cushioning agent and a second layer comprising a physical bJend of pressure sensitive multiparticulate and cushioning agent.
Examples of suitable cushioning agents include, alone or in any combination acacia, alginic acid, calcium carbonate, calcium phosphate dibasic anhydrous, calcium phosphate dibasic dihydrate, calcium phosphate tribasic, carbomer, carboxymethylcellulose calcium, carboxymethylcellulose sodium, carrageenan, cellulose acetate, cellulose powdered, chitosan, citric acid, colloidal siiicon dioxide, croscarmelose sodium, crospovidone, dextrates, dextrine, dextrose, dicalcium phosphate, ethylcellulose, fructose, gelatin, glucose, glyceryl behenate, glyceryl palmitostearate, guar gum, hydroxyethyl cellulose, hydroxyethylmethyl cellulose, hydroxypropyl cellulose, hydroxypropylmethyl cellulose, kaolin, lactitol, lactose, lactose calcium carbonate, low substituted hydroxypropyl cellulose, magnesium aluminum silicate, magnesium carbonate, magnesium oxide, maltodextrin, maltose, mannitol, methylcellulose, microcrystalline cellulose, pregelatinized starch, polacrilin potassium, polydextrose, polyethylene oxide, polymethacrylates, powdered cellulose, polyvinyl pyrrolidone, line
cellulose, simethicone, sodium alginate, sodium bicarbonate, sodium chloride, sorbitol, sodium starch glycolate, starch, sucrose, sugar (e.g., compressible, confectioner's, spheres), talc, trehalose, xylitol, zein, crossl inked polyvinylpyrrolidone and low- substituted hydroxypropyl cellulose. Where a layer of cushioning agent is employed, it is present in sufficient thickness to provide a cushioning effect during manufacture such that the release profile of the active is not materially altered. Preferably at least one portion of a first layer of cushioning agent has a thickness of at least 500 micrometers preferably at least 800 micrometers. If a second layer of cushioning agent is employed, then such second layer suitably has at least one portion thereof having a thickness of at least 500 micrometers and preferably at least 800 micrometers. Where two or more cushioning layers are employed, they are suitably of approximately the same thickness although they may be of different thickness if desired.
The pharmaceutical active can fall within any solubility class known in the art. It may include, for example, at least one pharmaceutical active that is a very soluble, freely soluble, soluble, poorly soluble or insoluble pharmaceutically active material. Suitably, the active material has a solubility in water of 1 g in less than 1 g water, 1 g in 1 to 10 g water, 1 g active in 10 to 30 g water, 1 g active in 30 to 100 g water, 1 g active in 100 to 1,000 g water, 1 g active in 1 ,000 to 10,000 g water, and 1 g active in more than 10,000 g water.
Examples of suitable classes of pharmaceutical actives include an analgesic, antiangina, antianaemia, antibiotic, antiarrhythmic, antidiarrheal, antidiuretic, antidepressant, antiemetic, antifungal, antirheumatic, antiviral, antiprotozoal, antihistamine, antihypertensive, anti-inflammatory, antimigraine, antinausea, antispasmodic, anxiolytic, beta blocker, calcium channel blocker, sedative, hypnotic, antipsychotic, bronchodilator, decongestant, cough expectorant, cough suppressant,
antiasthma drug, corticosteroid, actives for treatment of cough or common cold, muscle relaxant, erectile dysfunction active, motion sickness active, anti-HIV, anti-malaria actives, anti-cholesterol actives, respiratory actives, gastronintestinal actives, cardiovascular actives, antidiabetes actives, central nervous system actives, anti- infection actives, mucolytics, proton pump inhibitors and nasal decongestants
Examples of suitable actives include paracetamol, pseudoephedrine, acravastine, lamivudine, abacavir, pravastatin, Roziglitazone, ezetimibe, Clavulanate, sulfamethoxazole, benazepril, Valsartan, Irbesartan, Losartan, Dutasteride, tamsolusin, Atazanavir, ritonavir, propoxyphene, Hydrocodone, Metocarbamol, Memantine, Donepezil, Glyburide, Pioglytazone, Glimepiride, Benazepril, Torcetrapib, Eprosartan, Telmisartan, Olmesartan, Lopinavir, Emtricitabine, Tenofovir, Amprenavir, Tipranavir, Atovaquone, Proguanil, 5-aminosalicylic acid, 4-aminophthalic acid, Bismuth citrate, Bismuth subsalicylate, Montelukast, pseudoephedrine, Guaifenesin, ibuprofen, nifedipine, betamethasone acetate, methylprednisolone, dextromethorphan, cinnarazine, simvastatin, ciprofloxacin, glipizide, risperidone, glibenclamide, fenofibrate, isosorbide mononitrate, isosorbide dinitrate, acetazolamide, levothyroxine sodium, omeprazole, aspirin, codeine, dihydroergotamine, diazepam, theophylline, sildenafil citrate, vardenafil hydrochloride, amlodipine besylate, Zolpidem tartrate, acetaminophen, methocarbamol, ramipril, digoxin, enalapril maleate, fluoxetine hydrochloride, fexofenadine hydrochloride, olanzapine, methyldopa, hydrochlorothiazide, timolol maleate, alendronate sodium, thiabendazole, rofexocib, dicoflenac, bepridil hydrochloride, atorva statin hydrochloride, sertraline hydrochloride, famciclovir monohydrate, nabumetone, cimetidine, ketoprofen, etodolac, amiodarone hydrochloride, indomethacin, cefaclor, diltiazem, verapamil, felodipine, isradipine, nicardipine, prazosin, disopyramide, pentoxifilline, venlafaxine, alfuzosin, doxazosin, famotidine, ranitidine, pirenzipine, lansoprazole, loperamide, sulfasalazine, prednisolone, furosemide, amiloride, triamterene, verapamil, atenolol,
propranolol, captopril, glyceryl trinitrate, caffeine, aminophylline, cetirizine, loratadine, chlorpheniramine maleate, diphenhydramine, dothiepin, amitriptyline, phenelzine, paroxetine, fenfluramine, dimenhydrinate, ondansetron, domperidone, metoclopramide, tramadol, dihydrocodeine, pethidine, sumatriptan, amoxicillin, ampicillin, cefuroxime, cephalexin, tetracycline, erythromycin, co-trimoxazole, sulphadiazine, trimethoprim, nitrofurantoin, fluconazole, ketoconazole, acyclovir, zidovudine, chloroquine, mefloquin, metronidazole, metformin, chlorpropamide, ferrous sulphate, azapropazone, fenbufen, flurbiprofen, ketoprofen, naproxen, piroxicam, mefanamic acid, celecoxib, licofelone, tadalafil, mycophenolate, valgancyclovir, valacyclovir, sevelamer, metaxolone, nelfinavir, duranavir, tipranavir, levetiracetam, capecitabine, moxifloxacin, morphine, levofloxacin, clarithromycin, pregabalin, esomeprazole, quetiapine, efavirenz, oxcarbazepine, colesevelam, zileuton, nitazoxanide, clofibrate, praziquantel, sucralfate, cefprozil, indinavir, ganciclovir, oxaprozin, divalproex, cefadroxil, felbamate, potassium chloride, saquinavir, fosamprenavir, hydroxyurea, gabapentin, niacin, omega-3 acid ethyl esters, calcium acetate, progesterone, procainamide, delavirdine, ribavirin, propafenone, eprosartan, tocainide, tinidazole, choline magnesium trisalicylate, azithromycin, linezolid, lorazepam, oxazepam, lormetazepam, flunitrazepam, haloperidol, triptorelin, leuprorelin, lanreotide acetate, octreotide acetate, methylxanthin, tamsulosin, codeine hydrochloride, dextromoramide tartrate, ethymorphine hydrochloride, magnesium salicylate, methadone hydrochloride, oxycodone hydrochloride, sufentanil citrate, ephedrine, tramazoline hydrochloride, brompheniramine maleate, emedastine fumarate, and pharmaceutically or nutraceuticaly acceptable salts, acids, esters, isomers, and metabolites thereof.
Where more than one active material is present, the two or more actives may be from the same class or may be from different classes. Examples of combinations of active materials from different classes include an antibiotic in combination with one of a decongestant, an anti-inflammatory, a cough expectorant, a cough suppressant or an
active for treatment of cough or common cold, a proton pump inhibitor, anti-hypertension and anti-cholesterol actives.
Examples of classes where two or more active materials from one class may suitably be employed include respiratory actives, gastrointestinal actives, cardiovascular actives, antidiabetes actives, central nervous system actives, anti- infection actives, anti-viral actives, analgesics, anti-inflammatory actives, antibiotics, cough suppressants, expectorants, mucolytics, and nasal decongestants, anti-HIV , anti- malaria actives.
Examples of particular combinations of active materials include: Paracetamol and Caffeine; Aspirin and paracetamol; Paracetamol and pseudoephedrine; Paracetamol and phenylephrine; lbuprofen and codeine; lbuprofen and pseudoephedrine; Paracetamol and diphenhydramine; Acravistine and pseudoephedrine; Paracetamol and dextromethorphan; Parcetamol and guaphenesin; Paracetamol, caffeine, aspirin; Aspirin and caffeine; Zidovudine, lamivudine and abacavir; Pravastatin and aspirin; Lamivudine and zidovudine; Roziglitazone and Metformin; Ezetimibe and fenofibrate; Amoxicillin and Clavulanate; Trimetoprim and sulfamethoxazole; Amlodipine and benazepril; Valsartan and Hydrochlorothiazide; lrbesartan and Hydrochlorothiazide; Losartan and Hydrochlorothiazide; Fenofibrate and Metformin; Abacavir and lamivudine; Dutasteride and tamsolusin; Atazanavir and ritonavir; Ritonavir and Saquinavir; Propoxyphene and paracetamol; Hydrocodone and paracetamol; tramadol and paracetamol; Metocarbamol and paracetamol; Memantine and Donepezil; Glyburide and Metformin; Pioglytazone and Metformin; Rosiglitazone and Glimepiride, Benazepril and Hydrochlorothiazide; Atorvastatin and Torcetrapib; Eprosartan and Hydrochlorothiazide; Amlodipine and Atorvastatin; Ezetimibe and Simvastatin; Telmisartan and Hydrochlorothiazide; Olmesartan and Hydrochlorothiazide; Lopinavir and Ritonavir; Emtricitabine and Tenofovir; Fosamprenavir and Ritonavir; Amprenavir and Ritonavir; Tipranavir and
Ritonavir; Atovaquone and Proguanil; Lansoprazole, Amoxicillin and Clarithromycin; Lansoprazole and Naproxen; 5-aminosalicylic acid, 4-aminophthalic acid; Clarithromycin, Ranitidine and Bismuth citrate; Bismuth subsalicylate, Metronidazole and Tetracycline; Montelukast and Loratadine; Fexofenadine and pseudoephedrine; Guaifenesin and pseudoephedrine.
The pressure sensitive multiparticulate may be coated if desired. Any conventional coating may be employed for example a sustained release coating, enteric coating, taste-masking coating, moisture barrier coating, pressure sensitive barrier coating, pressure insensitive barrier coating and oxygen barrier coating and combinations thereof. Examples of such coating formulations suitably include at least one of methacrylates, ' methylcellulose, ethylcellulose, polyvinyl alcohol, hydroxypropylmethyl cellulose, hydroxypropyl cellulose, polyvinylacetate phthalate, methacrylic acid polymers, methacrylic ester copolymers, aminoalkyl methacrylate copolymers, hydroxypropylmethyl cellulose, carrageenan, ethylcellulose, starch acetates, polyethyl acrylate, polymethyl methacrylate, polymethacrylic acid, polyethyl acrylate, albumen, carboxymethyl cellulose, carboxymethylcellulose sodium, cellulose acetate, cellulose acetate phthalate, cetyl alcohol, chitosan, collagen, dextrin, gelatin, liquid glucose, glyceryl behenate, hyaluronic acid, hydroxyethyl cellulose, hydroxyethyl methyl cellulose, hydroxypropyl cellulose, hypromellose phthalate, lactose, maltitol, maltodextrin, methylcellulose, polydextrose, polyethylene oxide, polyvinyl acetate phthalate, polyvinyl alcohol, polyvinyl pyrilidone, sodium starch glycolate, shellac, camauba wax, microcrystalline wax and zein, acethyltriethyl citrate, triethyl citrate, tributyl citrate, acetyltri butyl citrate, dibutyl sebacate, diethyl phthalate, polyethylene glycol, 1 ,2-propylene glycol, glyceryl triacetate, glycerol, sorbitol, citric acid, lactic acid, triacetin, titanium dioxide, aluminium lakes, iron oxides, talc, magnesium stearate, glycerol monostearate, microcrystalline cellulose, colloidal silicon dioxide,
precipitated silicon dioxide, magnesium Al silicate, crosslinked polyvinylpyrrolidone, starch, lactose, alginic acid, carboxymethylcellulose calcium, carboxymethylcellulose sodium, powdered cellulose, chitosan, croscarmellose sodium, crospovidone, guar gum, low-substitued hydroxypropyl cellulose, methylcellulose, polacrilin potassium, povidone, sodium alginate, sodium starch glycolate, starch, pregelatinized starch, simethicone emulsion, polysorbate, sodium carboxymethycellulose.
The film to be used to enrobe the present invention may be any film capable of enrobing the compacted fill materials without adversely impacting the desired dissolution profile. The film to be used may comprise water soluble components, water insoluble components or may comprise soluble and insoluble components in combination.
If desired, the film material may be designed to be fast, immediate, delayed, pulsatile or sustained release.
Preferably, the compacted fill material of the present invention is enrobed by a film comprising at least one water soluble polymer. Films generally useful in the present invention include those that are thermo formable and generally have a dissolution rate appropriate for the preparation of rapid release, preferably immediate release, solid forms of the invention. Examples of such water soluble polymers include cellulosic materials such as hydroxyethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose; polyvinyl alcohol; hydrocolloids such as carrageenan, alginate and pectin; and water soluble acrylates. Examples of water insoluble polymers include ethylcellulose, methacrylates and cellulose acetate. The films used in the invention may be gelatin free. The films may contain plasticizers such as lactic acid, citric acid, polyethylene glycol, sorbitol, glycerine, triethylcitrate, propylene glycol, phthalates, triglycerides, triacetin, tributylcitrate, etc. WO 2004/026284, WO 02/083779 and WO 03/095548 disclose further examples of films that may be used in the invention and such are incorporated herein by reference. Examples of films that may be used in the present
invention are available under the trade name XGEL UNO from BioTec Films LLC, Tampa, Florida, US. Films for use in the present invention may be made in a conventional manner. If desired, an adhesive and use thereof can be used to aid in sealing the films together. Suitable adhesive compositions include those set forth in WO 04/10337 and WO 04/103338 - both of which are incorporated herein by reference.
The solid forms of the present invention may be enrobed and prepared in accordance with WO 03/096963, WO 05/030115, WO 05/030116 and PCT/GB2005/001077 - all of which are incorporated herein by reference.
The solid form of the present invention may be prepared using any equipment capable of enrobing a compacted fill material for use in the present invention
The invention also provides for the use of a solid form according to the invention in which the active material comprises a pharmaceutical active in the manufacture of a medicament for treatment of the human or animal body by therapy.
A further aspect of the invention provides a solid form according to the invention in which the active material comprises a pharmaceutical active for use in a method of treatment of the human or animal body by therapy. In the Figures:
Figures 1 and 2 show microscope images of solid forms which have been coated. Figures 1 shows pellets according to the invention and Figure 2 shows damaged pellets presented for comparative purposes and are not according to the invention.
Figures 3 to 5 show the release profile of a pharmaceutical active from a solid form according to the invention.
The present invention is described by reference to the following illustrative examples. Unless otherwise indicated herein, all parts, percents, ratios and the like are by weight.
EXAMPLES
The following materials, methods and equipment were used in the Examples set forth below unless otherwise indicated.
MATERIALS
Table 1

o Fluid bed; Glatt; Type: GPCG 3 o Compaction simulator; ESH, oblong flat punches; o Solid form production apparatus as described in WO 05/030115; BioProgress; o Hardness tester; Erweka; TBH30MD o Dissolution bath; Hanson Virtual Instruments; SR8PLUS and SIP o Spectrophotometer; Hewlett Packard 8453 o Microscope; SWIFT Instruments International; Stereo zoom eight o Balances (Mettler Toledo AG104, Mettler PM4800, Sartorius BP21 OS) o Mixers (Silverson L4R, IKA-WERKE)
METHODS 1) Coating conditions: Equipment: Fluid bed coater (Wurster technique)
(a) Preparation of coating of pressure sensitive multiparticulates with Eudragit NE30D:
Table 2: Eudragit NE30D suspension formulation

Talc was homogenized in water using a homogenizer (Silverson L4, high shear mixer) for 10 minutes. The prepared suspension was added slowly into the Eudragit NE30D dispersion while stirring gently with a conventional low shear mixer. Finally the
suspension was passed through a 0.5 mm sieve. The suspension was continuously stirred during the application of the coating.
(b) Preparation of top Coating (applied on top of Eudragit polymer coating):
Table 3: LustreClear LC103 suspension formulation

The LustreClear LC103 was dispersed in water using propeller mixer (IKA-WERKE, low shear mixer) for 50 minutes.
( c) Controlled Release Coating method:
Coating with the Eudragit NE30D suspension was performed under the following conditions: Inlet temperature: 26°C; Outlet temperature: 22°C; Spray rate: 3.0-4.7 g/min; Air across bed: 0.30 kPa; Atomization pressure: 1.5 bar; Spraying time: 85 minutes. Targeted weight gain was 12%.
(d)Top coating with LustreClear LC103 method:
The coating with LustreClear LC103 was performed under the following conditions: Inlet temperature: 26°C; Outlet temperature: 25°C; Spray rate: 3.0 g/min; Air across bed: 0.35 kPa; Atomization pressure: 1.5 bar; Spraying time: 35 minutes; Drying time: 10 minutes in the fluid bed. Targeted weight gain was 1%.
(e) Curing:
Prior to curing coated pellets were blended with 0.5 % Aerosil 200. Pellets were cured for a curing time of 2 hours on a tray at an oven temperature of 45°C.
Note: coatings with Eudragit NE30D and LustreClear LC103 were both done on the same pellets in the same coater, one after the other. LustreClear LC 103 is a second coating applied onto the Eudragit film. Its purpose is to decrease the sticking properties of the pellets coated with Eudragit NE30D.
(f) Preparation of coating of pressure sensitive multiparticulates with Eudragit RS/ Eudragit RL polymer:
Table 4: Eudragit RS/RL suspension formulation

The talc was homogenized in water using a homogenizer (Silverson L4, high shear mixer) for 10 minutes. The prepared suspension was added slowly into the Eudragit RS/RL dispersion while stirring gently with a conventional stirrer. Finally the suspension was passed through a 0.5 mm sieve. The suspension was continuously stirred during the coating process.
(g) Preparation of top Coating applied on top of Eudragit polymer:
Table 5: LustreClear LC103 suspension formulation

The LustreClear LC103 was dispersed in water using propeller mixer (IKA-WERKE, low shear mixer) for 50 minutes.
(h) Coating method:
The coating with Eudragit NE30D suspension was performed under the following conditions: Inlet temperature: 37°C; Outlet temperature: 27°C; Spray rate: 2.0-3.8 g/min; Air across bed: 0.30 kPa; Atomization pressure: 1.5 bar; Spraying time: ~180 minutes. Targeted weight gain was 10%.
(i) Top coating with LustreClear LC103 method
The coating with LustreClear LC103 was performed under the following conditions: Inlet temperature: 410C; Outlet temperature: 290C; Spray rate: 3.0 g/min; Air across bed: 0.35 kPa; Atomization pressure: 1.5 bar; Spraying time: ~35 minutes; Drying time: 10 minutes. Targeted weight gain was 1%.
G) Curing:
Prior to curing coated pellets were blended with 0.5 % Aerosil 200. Pellets were cured for a curing time of 24 hours on a tray at an oven temperature of 45°C. Note: coatings with Eudragit RS/RL and LustreClear LC103 were both done, on the same pellets in the same coater, one after the other. LustreClear LC 103 is second coating applied onto the Eudragit film. Its purpose is to decrease the sticking properties of the pellets coated with Eudragit RS/RL.
2) Fill material compacts manufacture:
Fill material compacts were prepared by compressing the fill material in an ESH compaction simulator (ESH Testing Ltd, Dudley, UK) with oblong flat punches having a dimension of 16.775 x 7.449 millimeters. The upper punch speed was 100 mm/sec and the cycle time was 0.36 second. The upper punch displacement was adjusted to vary the compaction force. The maximum applied pressure was chosen for the pellets- cushioning agent blend in order to obtain solid forms with good tensile strength. In the case of tabletting of the pellet/cushioning agent blend the pressure applied was around 177 MPa. Low compaction examples used for the compacted fill material was undertaken at a pressure of around 4.4 MPa. This low compaction pressure was used for the compression of: pellets without cushioning agent, pellets with cushioning agent (pellets - cushioning agent blend and pellets - cushioning agent - pellets layers). The weight of solid forms produced at low compaction pressure was varied according to the individual experiment. It was typically in the range between 350 and 800 milligrams. Six compacts were prepared for dissolution testing. The upper punch force and compact weight were recorded. The compaction pressure reported was based on the upper punch force divided by the punch area (113.03 square millimeters). Compacts were stored in double plastic bags prior to subsequent testing.
Materials were filled into the compaction simulator dye manually. Two approaches to solid form manufacture were investigated: layering technique and blend dye loading.
(a) Layering technique: o A Layer of cushioning agent was loaded into the dye cavity of the compaction simulator or into the thermoformed cup of the enrobing Lab Scale Machine. o On the top of the cushioning agent layer a layer of the coated pellets was added.
o Finally the coated pellets layer is covered again by a second layer of cushioning agent.
(b) Blending technique: A measured amount of cushioning agent and coated pellets were loaded into the dye cavity. The blending was performed in the dye (manual blending).
3) Hardness tests:
The hardness of the compacted dosages was evaluated on an Erweka hardness tester.
4) Microscope observation:
For easier observation through the microscope the dosages were gently dismantled (broken a part without causing any damage to the coated pellets). (Figures 1 and 2 show examples of undamaged and damaged pellets the latter being damaged during production due to the application of the compressive force or due to the absence of a cushioning agent. During the microscope observation the compacted dosages forms were thoroughly observed and any damage that occurred on the pellet coating monitored. Those pellets where the coating appeared damaged were counted (manual counting of damaged pellets). The % damage to all pellets was calculated as follows:
[number of damaged pellets x pellet weight (mg)]
X (%) = x 100 (%)
[pellets layer weight (mg)]
Two-sample t-test: The two-sample t-test is used to determine if two population means are equal. A common application of this is to test if a new process or treatment is superior to a current process or treatment. (Montgomery D.C. (2001), Design and Analysis of Experiments, John Wiley & Sons, INC, 35-36, 640)
5) Manufacture of enrobed solid form:
Soluble films known as XGEL UNO and supplied by Bio Tec Films LLC were cut into strips 6 centimeters by 20 centimeters approximately. The lower and upper films had a thickness of about 120 microns. The lower film was heated sufficiently to thermoform under vacuum into dose cups about 3 millimeters in height to conform to cavities (7.5 millimeters width by 16.75 length millimeters) with the cavity depth determined by height- adjustable dose-shaped lower pistons within the stainless steel die. The film strip was placed over the die and brought in contact with a heated TEFLON® coated surface by means of upward vacuum. The film was then drawn into the stainless steel die cavities by inverting the vacuum to form a strip of twelve thermoformed dose cups with 3.0 millimeters separation between adjacent dose cups. Some unused portion of the filmstrip was cut and removed. The fill composition was dosed by volume using a Kinematics and controls dosing system. Solid forms were manufactured using the layering technique with three different fill layers: 1) 100 mg layer of Avicel PH200; 2) 150 mg layer of Theophylline pellets coated with Eudragit NE30D; 3) 100 mg layer of Avicel PH200.
The multiple doses were filled one on top of the others so as to form horizontal layers. Then the layered fill was lightly compacted in the dose cups with upper pistons, and the lower film was cut to separate the individual solid forms. The solid forms were then lifted by the lower pistons to expose a portion of the solid form sidewalls for application of the upper film to complete the enrobing of the solid form. An adhesive
composed of 5% Methocel E15LV Premium, 45% Benzyl alcohol and 50% Triacetin, was applied (by transfer roller) to the upper filmstrip on the side to be pressed against the exterior sidewall of the dose cup. The upper film was placed over the solid forms containing the compressed layered powder fill and the film was heated by contact with the heating element using upper vacuum. The heated upper film was formed around the solid forms using the lower vacuum enclosing the fill material within the solid form by overlapping the upper film onto the sidewall of the solid forms. The top film was cut to separate the completed enrobed solid forms and the unused film was removed. The solid forms were further sealed by forcing them through a heated die under low pressure so that the cut film overlapping the sides was pressed smooth. All examples below used the apparatus set forth in WO 2005/030115,
Soluble HPMC containing films were used to enrobe the solid forms. Dissolution was determined according to United States Pharmacopeia LISP 24 with dissolution apparatus 1 , baskets. The sampling was undertaken at the following intervals of time: time zero then at intervals of 30 min, 30 min, 1 hour, 2 hours and every 2 hours up to the end point of the given dissolution.
Example 1
The theophylline pressure sensitive multiparticulates (pellets) were coated on the
Glatt GPC with the Eudragit NE30D polymer, formulation shown in table 2. The batch size was 1 kg. The Eudragit NE30D polymer weight gain was 4%. The top coating (LustreClear LC103) was applied to the theophylline pellets coated with Eudragit NE30D. LustreClear weight gain 1%. Total theophylline pellets weight gain after both coating was 13%.
The coated pellets were submitted to either compaction or enrobing (see methods described above) and the resulting solid form attributes were compared to the non-compacted (free) pellets (Sample #1 in Table 6). Sample 5 in Table 6 is a Comparative Example due to its high density and Sample 6 is a Comparative Example due to the absence of a cushioning agent.
Table 6:

CuA: cushioning agent ** In sample #5 the percentage of
CoP: coated pellets damaged pellets could not be evaluated
* In the samples 2 and 3 the quantity of because the compact was too hard and cushioning agent was composed of two could not be dismantled into initial layers each containing 100 mg of Avicel pressure sensitive multiparticulates
PH200 without further damage of film coat
Samples 1 and 6 in Table 6 did not contain any cushioning agent. Sample 1 did not undergo any compression and served as a control sample. During the compression of Samples 3-6 the compaction simulator settings were adjusted in accordance with the pressures set forth in Table 6. The compacts produced at the low compaction pressure
(4.4 - 5.6 MPa) had a very low hardness / tensile strength and in order to withstand the stress applied to the compact during post compaction handling a supporting enrobing film would be needed. Compaction pressures of 4.4 MPa would be expected to have a solid form density of lower than 1.1 g/ml, while a compaction pressure of 176.9 MPa (Sample 5) would be expected to provide an apparent density of greater than 1.1 g/ml. The drug release profiles for different samples set forth in Table 6 were evaluated as follows in Table 7 and illustrated in Figure 3
Table 7:
Time 0 0.5 1 2 4 6 8 10 12 14 16 18 20 22 24 (hours)
Sample 1. Uncompacted pellets mean 0.00 10.26 17.12 I 22.67 34.92 44.26 52.14 57.11 63.74 68.01 72.33 74.29 76.68 79.84 81.22
Sample 2. Enrobed solid form (150 mq pellets mean 0.00 9.60 14.55 19.27 34.15 44.41 52.72 59.10 64.18 68.36 72.07 76.70 79.34 82.07 84.38
Sample 3. Layering method (150 mg pellets) 4.4 MPa mean 0.00 1 10.36 17.60 24.30 37.71 47.95 54.41 60.19 64.77 67.68 70.96 75.13 77.88 80.13 82.00
Sample 4. Blend compaction (150 mg pellets) 4.4 MPa mean 0.00 11.15 17.95 25.16 36.41 45.47 54.60 60.40 65.51 69.69 73.99 75.29 76.92 78.25 79.97
Sample 5. Blend compaction (150 mg pellets) 176.9 MPa mean 0.00 11.19 25.74 40.55 65.52 80.79 87.04 92.05 95.89 96.78
K* X X X X 96.12
Sample 6. Compacted pellets (no cushioning agent) 5.3 MPa mean 0.00 28.50 40.81 49.81 66.23 71.02 76.40 80.18 82.23 84.40 86.30 87.68 88.73 88.93 90.44
A two-sample T-test was undertaken on the samples in Table 6 to show the equivalency or non-equivalency, with statistical significance of the different drug release profile evaluated versus the drug release profile of the non compacted pellets (Sample 1). The t test was performed on. the results obtained from the dissolution at 1, 4 and 12 hours. The t-test confidence limit is 95%. A to value between: -2.228 < t0 < 2.228 shows equivalence.
T — test results for Sample 1 (Table 6) were compared to Sample 3 (Table 6) and are set forth in Table 8.
Table 8:

At all three measured times Sample 3 showed statistical equivalency to Sample 1 in its drug release profile.
T -test results for Sample 1 (Table 6) were compared to Sample 2 (Table 6) and are set forth in Table 9.
Table 9:

At all three measured times Sample 2 showed statistical equivalency to Sample 1 in its drug release profile.
T - test results for Sample 1 (Table 6) were compared to Sample 4 (Table 6) and are set forth in Table 10.
Table 10:

At all three measured times Sample 4 showed statistical equivalency to Sample 1 in its drug release profile.
T — test results for Sample 1 (Table 6) were compared to Sample 5 (Table 6) and are set forth in Table 11. In this case a to value between: -2.365 < to < 2.365 show equivalence.
Table 11 :

At all three measured times Sample 5 showed statistical non-equivalency to Sample 1 in its drug release profile.
T - test results for Sample 1 (Table 6) were compared to Sample 6 (Table 6) and are set forth in Table 12. Table 12:

Table 13:

Example 2
Coated pellets used in this Example 2 are from the same batch as the pellets coated in Example 1.
Samples 1-3 in Table 14 are the same samples as Samples 1-3, respectively, in Table 6 of Example 1. In this example the pellets in Sample 4 were compressed using the layering technique at the compaction pressure of 4.4 MPa and by varying the pellets layer quantity. These compacts consisted of a higher amount of coated pellets than the compacts in Samples 2-3.
Table 14:

CuA: cushioning agent
CoP: coated pellets
* In the samples 2, 3 and 4 the quantity of cushioning agent was composed of two layers each containing 100 mg of Avicel PH200
** - Hardness
The coated pellets in Sample 1 (Table 14) did not undergo any compression and served as a control sample. Sample 2 (Table 14) was enrobed using the Enrobed solid form lab machine and Samples 3-4 (Table 14) were compressed using the compaction simulator. In samples 2-4 the cushioning agent was composed of two layers of Avicel PH200 (100 mg each). The amount of pellets in the layer varied from sample to sample. The compaction simulator settings were adjusted in order to obtain the compaction pressure of 4.4 MPa. Drug release profiles for different samples were evaluated using t- test analysis as set forth in Table 15. The data from Samples 1-3 in Table 14 is repeated from the same data in Table 7.
Table 15:

The obtained results are set forth in Figure 4.
A two-sample T-test was undertaken on Sample 4 (Table 14) to show the equivalency or non-equivalency,'' with statistical significance of the different drug release profile evaluated versus the drug release profile of the non compacted pellets. The t test was performed on the results obtained from the dissolution at 1 , 4 and 12 hours. The t- test confidence limit is 95%. A to value between: -2.228 < to < 2.228 shows equivalence. The t test data for Samples 2 and 3 showed statistical equivalence as compared to Sample 1 as noted above.
T - test results for Sample 1 (Table 14) were compared to Sample 4 (Table 14) and are set forth in Table 16. Table 16:

At all three measured times Sample 4 showed statistical equivalence to Sample 1 in its drug release profile. A summary is provided in Table 17. Table 17
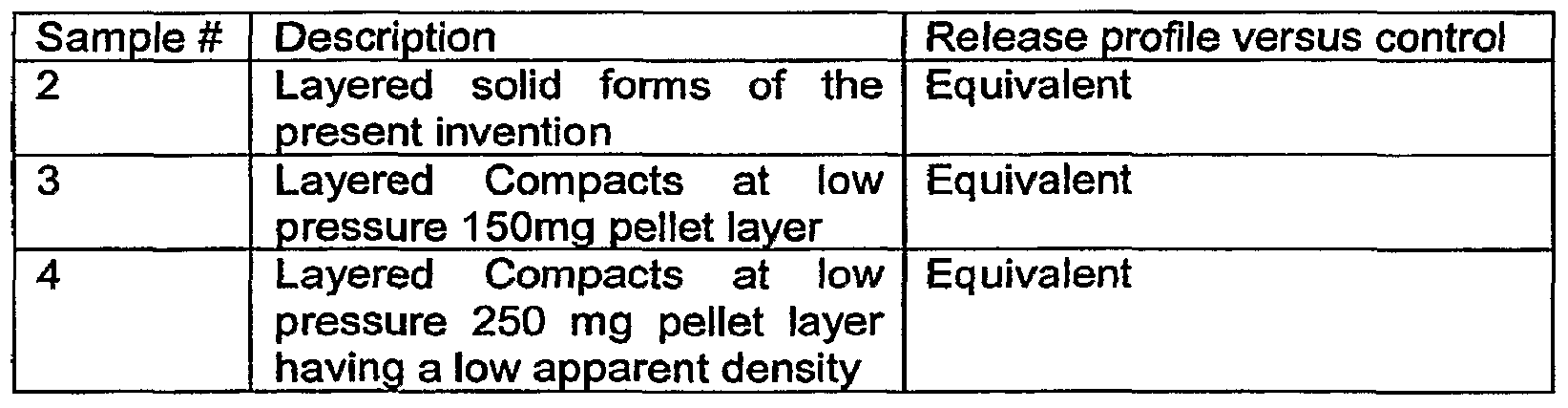
Example 3
Coated pellets used in this Example 3 are from the same batch as the pellets coated in Example 1. In examples 1 and 2 the cushioning agent used was Avicel PH200. This example shows the effectiveness of different cushioning agents in the present invention. As the effect on release profile has been shown to be directly related to the degree of pellet coating damage, microscopic examination was undertaken (as it has previously been shown that this attribute is predictive of the drug release). Table 18

In this example all dosages forms prepared and evaluated were compacts and the dosing method used was layering. Compaction was performed on Samples 1-4 in Table 18 using low pressure ( 4.4 MPa). During the compression the settings of the compaction simulator and the quantity of the pellets layer were kept unchanged. In order to obtain the compaction pressure of 4.4 MPa the quantity of the cushioning agent was adjusted accordingly. This adjustment in cushioning agent quantity was necessary because of different properties, e.g, density and consolidation of those three products evaluated. Table 18 shows the effectiveness of the inventive Samples 2-4 as compared to the degree of damaged pellets obtained for Sample 1.
Example 4
Theophylline pellets (batch size of 1 kg), were coated with an Eudragit RS/ Eudragit RL polymer blend (formulation methods described above in Table 4). The
pellets polymer weight gain was 6.6%. Top coating was LustreClear LC103 applied onto the pellets coated with Eudragit RS/ Eudragit RL. LustreClear weight gain was 1%. Total pellets weight gain after both coating was 11%w/w. In the following Table 19 the πon compacted pellets (Sample 1) were compared to the pellets compacted with cushioning agent (Avicel PH200; Sample 2) using the layering technique and to the pellets compacted at 4.4 MPa without cushioning (Sample 3).
Table 19

CoP: coated pellets CuA: cushioning agent
* In samples 2 the quantity of cushioning agent was composed of two layers each containing 100 mg of Avicel PH200
Sample 1 did not undergo any compression and served as a control sample. During the compression of sample 2 the compaction simulator settings were adjusted in order to obtain the desired compaction force.
Table 20: Drug release profiles of evaluated samples time 0 0.5 1 2 4 6 8 10 12 14 16 (hours)
Sample 1 Uncompacted pellets
Mean 0.0 0.32 0.83 1.60 14.43 28 .24 42. 89 57 .37 71.50 82.36 93.9 0 2
Sample : 2 Compacts 150 mg
Mean 0.0 3.32 3.06 4.09 16.38 28 .91 41. 79 53 .30 64.91 76.22 86.2 0 9
Sample '■ 3 Compacts No CuA
Mean 0.0 22.5 30.3 36.0 50.71 61 .28 71. 12 85 .64 91.45 96.63 97.3 0 3 5 6 6
Obtained drug release profiles are set forth in Figure 5.
A two-sample T-test was undertaken on Samples 1-3 to show the equivalency or non-equivalency, with statistical significance of the different drug release profile of Samples 2-3 evaluated versus the drug release profile of the non compacted pellets (Sample 1). The t test was performed on the results obtained from the dissolution at 4, 8 and 16 hours. In this case the first t-test was undertaken at the 4 hours time point because a significant lag time was observed in the release for the first 2 hours for all Samples 1-3 for this specific coating. Hence, a more significant data point is after the effects of this lag time have been taken into account.
T — test results for Sample 1 (Table 19) were compared to Sample 2 (Table 19) and are set forth in Table 21. In this case a t0 value between: -2.080 < to < 2.080 shows equivalency. Table 21

At all three measured times Sample 2 showed statistical equivalency to Sample 1 in its drug release profile.
T - test results for Sample 1 (Table 19) were compared to Sample 3 (Table 19) and are set forth in Table 22. In this case a to value between:: -2.120 < to < 2.120 shows equivalency. Table 22

Two samples out of three showed non-equivalency between the two drug release profiles evaluated for Sample 3 as compared to Sample 1.
While the invention has been described in detail and with reference to specific embodiments thereof, it will be apparent to one skilled in the art that various changes and modifications can be made therein without departing from the spirit and scope thereof.
Claims
1. A solid form comprising at least one film enrobing a compacted fill material wherein: i) the compacted fill material comprises: a) a pressure sensitive multiparticulate; and b) at least one cushioning agent; ii) the pressure sensitive multiparticulate and/or the cushioning agent comprises at least one active material; iii) the solid form has a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number
1216; iv) the compacted fill material has a density of at least 0.5 g/mf based on the total solid volume of the solid form and a tensile strength of less than 0.9
MPa.
2. A solid form comprising at least one film enrobing a compacted fill material wherein the compacted fill material comprises a pressure sensitive multiparticulate and at least one cushioning agent, the pressure sensitive multiparticulate and/or the cushioning agent comprising at least one active material, and the compacted fill material is selected from a pharmaceutical product, a food product, a veterinary product, a cosmetic, an appetite suppressant, a detergent product and a nutraceutical product, the said solid form shows a weight loss that is less than 1% during a 30 minutes USP friability test United States Pharmacopeia (USP) 29 Test Number 1216 (page 3046).
3. A solid form according to claim 1 or claim 2 wherein said compacted fill material is present in two or more discrete zones.
4. A solid form according to any one of the preceding claims wherein the compacted fill material comprises multiple layers, wherein a first layer comprises said cushioning agent and a second layer comprises said pressure sensitive multiparticulate.
5. A solid form according to claim 4 wherein the compacted fill material further comprises a third layer comprising a cushioning agent the arrangement of the layers being such that the said pressure sensitive multiparticulate layer is disposed between the two layers each comprising the said cushioning agent.
6. A solid form according to claim 1 or claim 2, wherein said compacted fill material comprises a physical blend comprising the pressure sensitive multiparticulate and the said cushioning agent.
7. A solid form according to claim 1 or claim 2, comprising at least one layer of said cushioning agent and a second layer comprising a physical blend of said pressure sensitive multiparticulate and said cushioning agent.
8. A solid form according to any one of the preceding claims, wherein said at least one cushioning agent comprises at least one of acacia, alginic acid, calcium carbonate, calcium phosphate dibasic anhydrous, calcium phosphate dibasic dihydrate, calcium phosphate tribasic, carbomer, carboxymethylcellulose calcium, carboxymethylcellulose sodium, carrageenan, cellulose acetate, cellulose powder, chitosan, citric acid, colloidal silicon dioxide, croscarmelose sodium, crospovidone, dextrates, dextrine, dextrose, dicalcium phosphate, ethytcellulose, fructose, gelatin, glucose, glyceryl behenate, glyceryl palmitostearate, guar gum, hydroxyethyl cellulose, hydroxyethylmethyl cellulose, hydroxypropyl cellulose, hydroxypropylmethyl cellulose, kaolin, lactitol, lactose, lactose calcium carbonate, low substituted hydroxypropyl cellulose, magnesium aluminum silicate, magnesium carbonate, magnesium oxide, maltodextrin, maltose, mannitol, methylcellulose, microcrystalline cellulose, pregelatinized starch, polacrilin potassium, polydextrose, polyethylene oxide, polymethacrylates, polyvinyl pyrrolidone, line cellulose, simethicone, sodium alginate, sodium bicarbonate, sodium chloride, sorbitol, sodium starch glycolate, starch, sucrose, sugar, talc, trehalose, xylitol, zein, crosslinked polyvinylpyrrolidone and low-substituted hydroxypropyl cellulose.
9. A solid form according to any one of the preceding claims, wherein said active material comprises at least one pharmaceutical active, nutraceutical active, food material, appetite suppressant, cosmetic component, detergent active, or industrial active material.
10 A solid form according to claim 9, wherein the said active material comprises at least one of a poorly soluble or insoluble pharmaceutically active material.
11. A solid form according to claim 9, wherein the said active material comprises at least one of a very soluble, freely soluble or soluble pharmaceutically active material.
12. A solid form according to any one of the preceding claims, wherein said pressure sensitive multiparticulate comprises at least one multiparticulate that is a sustained release composition comprising said active material.
13. A solid form according to any one of claims 1 to 12, wherein said pressure sensitive multiparticulate comprises at least one multiparticulate that is a delayed release composition comprising said active material.
14. A solid form according to any one of claims 1 to 12, wherein said pressure sensitive multiparticulates comprise at least one multiparticulate that is a pulsatile release composition comprising said active material.
15. A solid form according to any one of claims 1 to 12, wherein said pressure sensitive multiparticulate comprises at least one multiparticulate that is a fast release composition comprising said active material.
16. . A solid form according to any one of claims 1 to 12, wherein said pressure sensitive multiparticulate comprises at least one multiparticulate that is an immediate release composition comprising said active material.
17. A solid form according to any one of the preceding claims, wherein said active material comprises at least one of an analgesic, antiangina, antianaemia, antibiotic, antiarrhythmic, antidiarrheal, antidiuretic, antidepressant, antiemetic, antifungal, antirheumatic, antiviral, antiprotozoal, antihistamine, antihypertensive, anti-inflammatory, antimigraine, antinausea, antispasmodic, anxiolytic, beta blocker, calcium channel blocker, sedative, hypnotic, antipsychotic, bronchodilator, decongestant, cough expectorant, cough suppressant, antiasthma drug, corticosteroid, actives for treatment of cough or common cold, muscle relaxant, erectile dysfunction active, motion sickness active, anti-HIV, anti-malaria actives, anti-cholesterol actives, respiratory actives, gastrointestinal actives, cardiovascular actives, antidiabetes actives, central nervous system actives, anti-infection actives, mucolytics, and nasal decongestants.
"18. A solid form according to any one of the preceding claims, comprising at least two active materials wherein the active materials are selected from: i) an antibiotic in combination with a decongestant, an anti-inflammatory, a cough expectorant, a cough suppressant or an active for treatment of cough or common cold; ii) an anti-HIV and anti-malaria active material; iii) an anti-hypertension and anti-cholesterol active material and iv) two or more active materials from the same class of active materials, the class being selected from respiratory actives, gastronintestinal actives, cardiovascular actives, antidiabetes actives, central nervous system actives, anti-infection actives, anti-viral actives, analgesics, anti-inflammatory actives, antibiotics, cough suppressants, expectorants, mucolytics, and nasal decongestants.
19. A solid form according to any one of the preceding claims, wherein the said at least one active material comprises paracetamol, pseudoephedrine, acravastine, lamivudine, abacavir, pravastatin, Roziglitazone, ezetimibe, Clavulanate, sulfamethoxazole, benazepril, Valsartan, Irbesartan, Losartan, Dutasteride, tamsolusin, Atazanavir, ritonavir, propoxyphene, Hydrocodone, Metocarbamol, Memantine, Donepezil, Glyburide, Pioglytazone, Glimepiride, Benazepril, Torcetrapib, Eprosartan, Telmisartaπ, Olmesartan, Lopinavir, Emtricitabine, Tenofovir, Amprenavir, Tipranavir, Atovaquone, Proguanil, 5-aminosalicylic acid, 4-aminophthalic acid, Bismuth citrate, Bismuth subsalicylate, Montelukast, PSE, Guaifenesin, ibuprofen, nifedipine, betamethasone acetate, methylprednisolone, dextromethorphan, cinnarazine, simvastatin, ciprofloxacin, glipizide, risperidone, glibenciamide, fenofibrate, isosorbide mononitrate, isosorbide dinitrate, acetazolamide, levothyroxine sodium, omeprazole, aspirin, codeine, dihydroergotamine, diazepam, theophylline, sildenafil citrate, vardenafil hydrochloride, amlodipine besylate, Zolpidem tartrate, acetaminophen, methocarbamol, ramipril, digoxin, enalapril maleate, fluoxetine hydrochloride, fexofenadine hydrochloride, olanzapine, methyldopa, hydrochlorothiazide, timolol mateate, alendronate sodium, thiabendazole, rofexocib, dicoflenac, bepridil hydrochloride, atorvastatin hydrochloride, sertraline hydrochloride, famciclovir monohydrate, nabumetone, cimetidine, ketoprofen, etodolac, amiodarone hydrochloride, indomethacin, cefaclor, diltiazem, verapamil, felodipine, isradipine, nicardipine, prazosin, disopyramide, pentoxifilline, venlafaxine, alfuzosin, doxazosin, famotidine, ranitidine, pirenzipine, lansoprazole, loperamide, sulfasalazine, prednisolone, furosemide, amiloride, triamterene, verapamil, atenolol, propranolol, captopril, glyceryl trinitrate, caffeine, aminophylline, cetirizine, loratadine, chlorpheniramine maleate, diphenhydramine, dothiepin, amitriptyline, phenelzine, paroxetine, fenfluramine, dimenhydrinate, ondansetron, domperidone, metoclopramide, tramadol, dihydrocodeine, pethidine, sumatriptan, amoxicillin, ampicillin, cefuroxime, cephalexin, tetracycline, erythromycin, co-trimoxazoie, sulphadiazine, trimethoprim, nitrofurantoin, fluconazole, ketoconazole, acyclovir, zidovudine, chloroquine, mefloquin, metronidazole, metformin, chlorpropamide, ferrous sulphate, azapropazone, fenbufen, flurbiprofen, ketoprofen, naproxen, piroxicam, mefanamic acid, celecoxib, licofelone, tadalafϊl, mycophenolate, valgancyclovir, valacyclovir, sevelamer, metaxolone, nelfinavir, duranavir, tipranavir, levetiracetam, capecitabine, moxifloxacin, moφhine, levofloxacin, clarithromycin, pregabalin, esomeprazole, quetiapine, efavirenz, oxcarbazepine, colesevelam, zileuton, nitazoxanide, clofibrate, praziquantel, sucralfate, cefprozil, indinavir, ganciclovir, oxaprozin, divalproex, cefadroxil, felbamate, potassium chloride, saquinavir, fosamprenavir, hydroxyurea, gabapentin, niacin, omega-3 acid ethyl esters, calcium acetate, progesterone, procainamide, delavirdine, ribavirin, propafenone, eprosartan, tocainide, tinidazole, choline magnesium trisalicylate, azithromycin, linezolid, lorazepam, oxazepam, lormetazepam, flunitrazepam, haloperidol, triptorelin, leuprorelin, lanreotide acetate, octreotide acetate, methylxanthin, tamsulosin, codeine hydrochloride, dextromoramide tartrate, ethymorphine hydrochloride, magnesium salicylate, methadone hydrochloride, oxycodone hydrochloride, sufentanil citrate, ephedrine, tramazoline hydrochloride, brompheniramine maleate, emedastiπe fumarate, and pharmaceuticaly or nutraceuticaly acceptable salts, acids, esters, isomers, and metabolites thereof.
20. A solid form according to any one of the preceding claims, comprising at least two active materials wherein the active materials are selected from: Paracetamol and Caffeine; Aspirin and paracetamol; Paracetamol and pseudoephedrine; Paracetamol and phenylephrine; lbuprofen and codeine; lbuprofen and pseudoephedrine; Paracetamol and diphenhydramine; Acravistine and pseudoephedrine; Paracetamol and dextromethorphan; Parcetamol and guaphenesin; Paracetamol, caffeine, aspirin; Aspirin and caffeine; Zidovudine, lamivudine and abacavir; Pravastatin and aspirin; Lamivudine and zidovudine; Roziglitazone and Metformin; Ezetimibe and fenofibrate; Amoxicillin and Clavulanate; Trimetoprim and sulfamethoxazole; Amlodipine and benazepril; Valsartan and Hydrochlorothiazide; lrbesartan and Hydrochlorothiazide; Losartan and Hydrochlorothiazide; Fenofibrate and Metformin; Abacavir and lamivudine; Dutasteride and tamsolusin; Atazanavir and ritonavir; Ritonavir and Saquinavir; Propoxyphene and paracetamol; Hydrocodone and paracetamol; tramadol and paracetamol; Metocarbamol and paracetamol; Memantine and Donepezil; Glyburide and Metformin; Pioglytazone and Metformin; Rosiglitazone and Glimepiride, Benazepril and Hydrochlorothiazide; Atorvastatiπ and Torcetrapib; Eprosartan and Hydrochlorothiazide; Amlodipine and Atorvastatin; Ezetimibe and Simvastatin; Telmisartan and Hydrochlorothiazide; Olmesartan and Hydrochlorothiazide; Lopinavir and Ritonavir; Emtricitabine and Tenofovir; Fosamprenavir and Ritonavir; Amprenavir and Ritonavir; Tipranavir and Ritonavir; Atovaquone and Proguanil; Lansoprazole, Amoxicillin and Clarithromycin; Lansoprazole and Naproxen; 55- aminosalicylic acid, 4-aminophthalic acid; Clarithromycin, Ranitidine and Bismuth citrate; Bismuth subsalicylate, Metronidazole and Tetracycline; Montelukast and Loratadine; Fexofenadine and pseudoephedrine; Guaifenesin and pseudoephedrine.
21. A solid form according to any one of the preceding claims, wherein said cushioning agent comprises a pharmaceutical active material.
22. A solid form according to any one of the preceding claims, wherein said pressure sensitive multiparticulate comprises at least one of pellets, granules, spheres, microspheres, freeze dried material or crystals.
23. A solid form according to any one of the preceding claims wherein at least one of said pressure sensitive multiparticulates is coated and said coating comprises at least one of a sustained release coating, enteric coating, taste-masking coating, moisture barrier coating, pressure sensitive barrier coating, pressure insensitive barrier coating and oxygen barrier coating.
24. A solid form according to claim 23, wherein the coating comprises at least one of methacrylates, methylcellulose, ethylcellulose, polyvinyl alcohol, hydroxypropylmethyl cellulose, hydroxypropyl cellulose, polyvinylacetate phthalate, methacrylic acid polymers, methacrylic ester copolymers, aminoalkyl methacrylate copolymers, hydroxypropylmethyl cellulose, carrageenan, ethylcellulose, starch acetates, polyethyl acrylate, polymethyl methacrylate, polymethacrylic acid, polyethyl acrylate, albumen, carboxymethyl cellulose, carboxymethylcellulose sodium, cellulose acetate, cellulose acetate phthalate, cetyl alcohol, chitosan, collagen, dextrin, gelatin, liquid glucose, glyceryl behenate, hyaluronic acid, hydroxyethyl cellulose, hydroxyethylmethyl cellulose, hydroxypropyl cellulose, hypromellose phthalate, lactose, maltitol, maltodextrin, methylcellulose, polydextrose, polyethylene oxide, polyvinyl acetate phthalate, polyvinyl alcohol, polyvinyl pyrilidone, sodium starch glycolate, shellac, carnauba wax, microcrystalline wax and zein, acethyltriethyl citrate, triethyl citrate, tributyl citrate, acetyltributyl citrate, dibutyl sebacate, diethyl phthalate, polyethylene glycol, 1,2- propylene glycol, glyceryl triacetate, glycerol, sorbitol, citric acid, lactic acid, triacetin, titanium dioxide, aluminium lakes, iron oxides, talc, magnesium stearate, glycerol monostearate, microcrystalline cellulose, colloidal silicon dioxide, precipitated silicon dioxide, magnesium Al silicate, crosslinked polyvinylpyrrolidone, starch, lactose, alginic acid, carboxymethylcellulose calcium, carboxymethylcellulose sodium, powdered cellulose, chitosan, croscarmellose sodium, crospovidone, guar gum, low-substitued hydroxypropyl cellulose, methylcellulose, polacrilin potassium, povidone, sodium alginate, sodium starch glycolate, starch, pregelatinized starch, simethicone emulsion, polysorbate, sodium carboxymethycellulose.
25. A solid form according to any one of the preceding claims, wherein said solid form further comprises a pressure-insensitive material.
26. A solid form according to any one of the preceding claims wherein the cushioning agent is present as a layer having a thickness of at least 500 micrometers.
27. A solid form according to claim 26, wherein at least one portion of the layer of cushioning agent has a thickness of at least 800 micrometers.
28. A solid form according to any one of the preceding claims comprising a first layer comprising a cushioning agent and a second layer comprising a pressure sensitive multiparticulate and a cushioning agent.
29. A solid form according to any one of the preceding claims wherein the tensile strength of the compacted fill material is less than 0.2 MPa.
30. A solid form according to any one of the preceding claims wherein said pressure sensitive multiparticulates are prepared by granulation, freeze drying, spray drying, roller compaction, lyophilization, extrusion, spheronization or milling.
31. A solid form according to any one of the preceding claims, wherein the film enrobing the compacted fill material is a water-soluble film.
32. A solid form according to any one of claims 1 to 31 in which the active material comprises a pharmaceutical active for use in a method of treatment of the human or animal body by therapy.
33. Use of a solid form according to any one of claims 1 to 32 in which the active material comprises a pharmaceutical active, in the manufacture of a medicament for use in a method of treatment of the human or animal body by therapy.
34 A method of making a method of making a solid form comprising at least one film enrobing a compacted fill material, the solid form having a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number 1216 and the compacted fill material having a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa, the said method comprising: i) providing a first film shaped to define an interior volume for holding a compacted fill material and having an open end; ii) depositing a fill material in the interior volume the fill material comprising a pressure sensitive multiparticulate and a cushioning agent and wherein at least one of the said multiparticulate and the cushioning agent comprises at least one active material; iii) applying pressure to the fill material so as to compact the fill material; iv) applying a second film over the said open end to close the said open end; and v) sealing the first and second films together to enrobe the compacted fill material and provide the solid form.
35. A method of making a solid form comprising at least one film enrobing a compacted fill material, the solid form having a weight loss that is less than 1% during a 30 minute friability test in accordance with United States Pharmacopeia 29 Test Number 1216 and the compacted fill material having a density of at least 0.5 g/ml based on the total solid volume of the solid form and a tensile strength of less than 0.9 MPa, the said method comprising: i) providing a first film shaped to define an interior volume for holding a compacted fill material and having an open end; ii) depositing via the open end a first zone of a first fill material in the interior volume; iii) depositing a second zone of a second fill material in the interior volume such that the interior volume comprises two zones of fill material wherein one of the first or second fill materials comprises a pressure sensitive multiparticulate and the other of the said first or second fill materials comprises a cushioning agent and wherein at least one of the said multiparticulate and the cushioning agent comprises at least one active material; iv) applying pressure to the fill material so as to compact the at least two zones of fill material v) applying a second film over the said open end to close the said open end; and vii) sealing the first and second films together to enrobe the compacted fill material and provide the solid form.
36. A method of making a solid form according to claim 35 in which the first fill material comprises a cushioning agent, the second fill material comprises an optionally coated multiparticulate and which method further comprises depositing in a third zone a third fill material comprising a cushioning agent which may be the same or different to the first fill material.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2007/011707 WO2008140459A1 (en) | 2007-05-16 | 2007-05-16 | Solid form |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2007/011707 WO2008140459A1 (en) | 2007-05-16 | 2007-05-16 | Solid form |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008140459A1 true WO2008140459A1 (en) | 2008-11-20 |
Family
ID=38665270
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/011707 WO2008140459A1 (en) | 2007-05-16 | 2007-05-16 | Solid form |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008140459A1 (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2343055A1 (en) | 2009-12-22 | 2011-07-13 | Sanovel Ilac Sanayi ve Ticaret A.S. | Pharmaceutical compositions of pregabalin |
| EP2389934A1 (en) | 2010-05-25 | 2011-11-30 | Sanovel Ilac Sanayi ve Ticaret A.S. | Controlled-Release Tablet Formulations of Pregabalin |
| EP2389933A1 (en) | 2010-05-25 | 2011-11-30 | Sanovel Ilac Sanayi ve Ticaret A.S. | Controlled-Release Pregabalin Compositions |
| EP2389935A1 (en) | 2010-05-25 | 2011-11-30 | Sanovel Ilac Sanayi ve Ticaret A.S. | Controlled-Release oral Solution Formulations of Pregabalin |
| EP2409701A1 (en) | 2010-07-19 | 2012-01-25 | Sanovel Ilac Sanayi ve Ticaret A.S. | Prasugrel granules with improved stability |
| CN101744726B (en) * | 2010-01-18 | 2012-12-26 | 北京东兴堂科技发展有限公司 | Sealing technology of plant capsule |
| CN103120803A (en) * | 2012-12-29 | 2013-05-29 | 钟春燕 | Preparation method of bacterial cellulose composite chitosan moist antimicrobial dressing |
| CN103212106A (en) * | 2013-04-18 | 2013-07-24 | 钟春燕 | Method for preparing bacterial cellulose slow-release analgesia dressing |
| US8501816B2 (en) | 2010-10-12 | 2013-08-06 | Cerecor, Inc. | Antitussive compositions comprising memantine |
| US8580302B2 (en) | 2000-11-20 | 2013-11-12 | Warner Chilcott Company, Llc | Pharmaceutical dosage form with multiple coatings for reduced impact of coating fractures |
| US8637540B2 (en) | 2003-11-26 | 2014-01-28 | Acura Pharmaceuticals | Compositions for deterring abuse of opioid containing dosage forms |
| US8901113B2 (en) | 2009-09-30 | 2014-12-02 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse |
| US8920838B2 (en) | 2006-08-03 | 2014-12-30 | Horizon Pharma Ag | Delayed-release glucocorticoid treatment of rheumatoid disease |
| CN104257665A (en) * | 2014-09-01 | 2015-01-07 | 南京正大天晴制药有限公司 | Oral solid preparation of dutasteride and preparation method thereof |
| US9101636B2 (en) | 2012-11-30 | 2015-08-11 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| CN105796524A (en) * | 2016-03-29 | 2016-07-27 | 南京多宝生物科技有限公司 | Aspirin enteric-coated tablet |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| CN106109471A (en) * | 2016-07-04 | 2016-11-16 | 浙江爱诺药业股份有限公司 | A kind of preparation method improving irbesartan and hydrochlorthiazide sheet uniformity of dosage units |
| CN106727357A (en) * | 2017-01-19 | 2017-05-31 | 杭州华东医药集团新五丰药业有限公司 | Acyclovir granule and preparation method thereof |
| KR20170070280A (en) * | 2011-04-08 | 2017-06-21 | 브라코 이미징 에스.피.에이. | Process for the preparation of a sulfated derivative of 3,5-diiodo-o-[3-iodophenyl]-l-tyrosine |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| CN107157940A (en) * | 2017-07-07 | 2017-09-15 | 青岛农业大学 | A kind of Lung targeting sulphuric acid cephalosporium quinol gelatine microsphere and preparation method thereof |
| US9890116B2 (en) | 2002-11-13 | 2018-02-13 | Bracco Imaging S.P.A. | Process for the preparation of a sulfated derivative of 3,5-diiodo-O-[3-iodophenyl]-L-tyrosine |
| JP2018118936A (en) * | 2017-01-27 | 2018-08-02 | 日本ケミファ株式会社 | Composition for preventing breakage of enteric layer |
| US10076494B2 (en) | 2016-06-16 | 2018-09-18 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| CN109432029A (en) * | 2018-02-01 | 2019-03-08 | 海南天煌制药有限公司 | A kind of tenofovir disoproxil fumarate ester tablets and preparation method thereof |
| US10238615B2 (en) | 2002-11-13 | 2019-03-26 | Bracco S.P.A. | 3,5,3′-triiodothyronine sulfate as thyromimetic agent and pharmaceutical formulations thereof |
| CN110381926A (en) * | 2017-03-09 | 2019-10-25 | 德维科制药瑞士公司 | Novel dosage forms |
| CN110917137A (en) * | 2019-11-26 | 2020-03-27 | 江南大学 | Preparation method of ultrastable pickering emulsion with synergistic stability of prolamin nanoparticles and starch nanoparticles |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| US11077055B2 (en) | 2015-04-29 | 2021-08-03 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
| US11103581B2 (en) | 2015-08-31 | 2021-08-31 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US11179331B1 (en) | 2020-04-21 | 2021-11-23 | Cure Pharmaceutcai Holding Corp | Oral soluble film containing sildenafil citrate |
| CN115247003A (en) * | 2022-08-11 | 2022-10-28 | 安徽凤阳赛吉元无机材料有限公司 | Novel white carbon black and preparation method thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4353887A (en) * | 1979-08-16 | 1982-10-12 | Ciba-Geigy Corporation | Divisible tablet having controlled and delayed release of the active substance |
| EP0377439A2 (en) * | 1989-01-03 | 1990-07-11 | Sterling Winthrop Inc. | Controlled-release, low-dose aspirin |
| US20020155153A1 (en) * | 1996-01-08 | 2002-10-24 | Astrazeneca Ab. | Oral pharmaceutical dosage forms comprising a proton pump inhibitor and a NSAID |
| US20060003005A1 (en) * | 2004-07-02 | 2006-01-05 | Bruce Cao | Tablet for pulsed delivery |
-
2007
- 2007-05-16 WO PCT/US2007/011707 patent/WO2008140459A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4353887A (en) * | 1979-08-16 | 1982-10-12 | Ciba-Geigy Corporation | Divisible tablet having controlled and delayed release of the active substance |
| EP0377439A2 (en) * | 1989-01-03 | 1990-07-11 | Sterling Winthrop Inc. | Controlled-release, low-dose aspirin |
| US20020155153A1 (en) * | 1996-01-08 | 2002-10-24 | Astrazeneca Ab. | Oral pharmaceutical dosage forms comprising a proton pump inhibitor and a NSAID |
| US20060003005A1 (en) * | 2004-07-02 | 2006-01-05 | Bruce Cao | Tablet for pulsed delivery |
Non-Patent Citations (1)
| Title |
|---|
| M.E. AULTON: "Pharmaceutics: The science of dosage form design, 2nd edition", 2002, CHURCHILL LIVINGSTONE, LONDON, XP002459291 * |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9089492B2 (en) | 2000-11-20 | 2015-07-28 | Warner Chilcott Company, Llc | Pharmaceutical dosage form with multiple coatings for reduced impact of coating fractures |
| US8580302B2 (en) | 2000-11-20 | 2013-11-12 | Warner Chilcott Company, Llc | Pharmaceutical dosage form with multiple coatings for reduced impact of coating fractures |
| US10238615B2 (en) | 2002-11-13 | 2019-03-26 | Bracco S.P.A. | 3,5,3′-triiodothyronine sulfate as thyromimetic agent and pharmaceutical formulations thereof |
| US9890116B2 (en) | 2002-11-13 | 2018-02-13 | Bracco Imaging S.P.A. | Process for the preparation of a sulfated derivative of 3,5-diiodo-O-[3-iodophenyl]-L-tyrosine |
| US8822489B2 (en) | 2003-11-26 | 2014-09-02 | Acura Pharmaceuticals | Abuse deterrent compositions and methods of making same |
| US9492443B2 (en) | 2003-11-26 | 2016-11-15 | Acura Pharmaceuticals, Inc. | Abuse deterrent compositions and methods of making same |
| US8637540B2 (en) | 2003-11-26 | 2014-01-28 | Acura Pharmaceuticals | Compositions for deterring abuse of opioid containing dosage forms |
| US9504699B2 (en) | 2006-08-03 | 2016-11-29 | Hznp Limited | Delayed-release glucocorticoid treatment of rheumatoid disease |
| US8920838B2 (en) | 2006-08-03 | 2014-12-30 | Horizon Pharma Ag | Delayed-release glucocorticoid treatment of rheumatoid disease |
| US10155044B2 (en) | 2009-09-30 | 2018-12-18 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse |
| US8901113B2 (en) | 2009-09-30 | 2014-12-02 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse |
| EP2343055A1 (en) | 2009-12-22 | 2011-07-13 | Sanovel Ilac Sanayi ve Ticaret A.S. | Pharmaceutical compositions of pregabalin |
| CN101744726B (en) * | 2010-01-18 | 2012-12-26 | 北京东兴堂科技发展有限公司 | Sealing technology of plant capsule |
| EP2389935A1 (en) | 2010-05-25 | 2011-11-30 | Sanovel Ilac Sanayi ve Ticaret A.S. | Controlled-Release oral Solution Formulations of Pregabalin |
| EP2389933A1 (en) | 2010-05-25 | 2011-11-30 | Sanovel Ilac Sanayi ve Ticaret A.S. | Controlled-Release Pregabalin Compositions |
| EP2389934A1 (en) | 2010-05-25 | 2011-11-30 | Sanovel Ilac Sanayi ve Ticaret A.S. | Controlled-Release Tablet Formulations of Pregabalin |
| EP2409701A1 (en) | 2010-07-19 | 2012-01-25 | Sanovel Ilac Sanayi ve Ticaret A.S. | Prasugrel granules with improved stability |
| US8501816B2 (en) | 2010-10-12 | 2013-08-06 | Cerecor, Inc. | Antitussive compositions comprising memantine |
| KR20170070280A (en) * | 2011-04-08 | 2017-06-21 | 브라코 이미징 에스.피.에이. | Process for the preparation of a sulfated derivative of 3,5-diiodo-o-[3-iodophenyl]-l-tyrosine |
| US10457635B2 (en) | 2011-04-08 | 2019-10-29 | Bracco Imaging S.P.A. | Process for the preparation of a sulfated derivative of 3,5-diiodo-o-[3-iodophenyl]-l-tyrosine |
| KR101865883B1 (en) * | 2011-04-08 | 2018-06-08 | 브라코 이미징 에스.피.에이. | Process for the preparation of a sulfated derivative of 3,5-diiodo-o-[3-iodophenyl]-l-tyrosine |
| US9320796B2 (en) | 2012-11-30 | 2016-04-26 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US9101636B2 (en) | 2012-11-30 | 2015-08-11 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US10688184B2 (en) | 2012-11-30 | 2020-06-23 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US11857629B2 (en) | 2012-11-30 | 2024-01-02 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US10441657B2 (en) | 2012-11-30 | 2019-10-15 | Abuse Deterrent Pharmaceuticals, Llc | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| CN103120803A (en) * | 2012-12-29 | 2013-05-29 | 钟春燕 | Preparation method of bacterial cellulose composite chitosan moist antimicrobial dressing |
| CN103212106A (en) * | 2013-04-18 | 2013-07-24 | 钟春燕 | Method for preparing bacterial cellulose slow-release analgesia dressing |
| US10639281B2 (en) | 2013-08-12 | 2020-05-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10792254B2 (en) | 2013-12-17 | 2020-10-06 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| CN104257665A (en) * | 2014-09-01 | 2015-01-07 | 南京正大天晴制药有限公司 | Oral solid preparation of dutasteride and preparation method thereof |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| US11077055B2 (en) | 2015-04-29 | 2021-08-03 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
| US11103581B2 (en) | 2015-08-31 | 2021-08-31 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| CN105796524A (en) * | 2016-03-29 | 2016-07-27 | 南京多宝生物科技有限公司 | Aspirin enteric-coated tablet |
| CN105796524B (en) * | 2016-03-29 | 2019-01-01 | 山西兰花药业股份有限公司 | A kind of aspirin enteric coated tablet |
| US10076494B2 (en) | 2016-06-16 | 2018-09-18 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| US10835488B2 (en) | 2016-06-16 | 2020-11-17 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| CN106109471A (en) * | 2016-07-04 | 2016-11-16 | 浙江爱诺药业股份有限公司 | A kind of preparation method improving irbesartan and hydrochlorthiazide sheet uniformity of dosage units |
| CN106727357B (en) * | 2017-01-19 | 2020-09-18 | 杭州益品新五丰药业有限公司 | Acyclovir granules and preparation method thereof |
| CN106727357A (en) * | 2017-01-19 | 2017-05-31 | 杭州华东医药集团新五丰药业有限公司 | Acyclovir granule and preparation method thereof |
| JP2018118936A (en) * | 2017-01-27 | 2018-08-02 | 日本ケミファ株式会社 | Composition for preventing breakage of enteric layer |
| JP7064683B2 (en) | 2017-01-27 | 2022-05-11 | 日本ケミファ株式会社 | Composition for preventing damage to the enteric layer |
| CN110381926A (en) * | 2017-03-09 | 2019-10-25 | 德维科制药瑞士公司 | Novel dosage forms |
| CN107157940A (en) * | 2017-07-07 | 2017-09-15 | 青岛农业大学 | A kind of Lung targeting sulphuric acid cephalosporium quinol gelatine microsphere and preparation method thereof |
| CN109432029A (en) * | 2018-02-01 | 2019-03-08 | 海南天煌制药有限公司 | A kind of tenofovir disoproxil fumarate ester tablets and preparation method thereof |
| CN110917137B (en) * | 2019-11-26 | 2021-08-10 | 江南大学 | Preparation method of Pickering emulsion with synergistic and stable prolamin nanoparticles and starch nanoparticles |
| CN110917137A (en) * | 2019-11-26 | 2020-03-27 | 江南大学 | Preparation method of ultrastable pickering emulsion with synergistic stability of prolamin nanoparticles and starch nanoparticles |
| US11179331B1 (en) | 2020-04-21 | 2021-11-23 | Cure Pharmaceutcai Holding Corp | Oral soluble film containing sildenafil citrate |
| CN115247003A (en) * | 2022-08-11 | 2022-10-28 | 安徽凤阳赛吉元无机材料有限公司 | Novel white carbon black and preparation method thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20080311162A1 (en) | Solid form | |
| WO2008140459A1 (en) | Solid form | |
| US20080286344A1 (en) | Solid form | |
| US20080286343A1 (en) | Solid form | |
| US20080014228A1 (en) | Solid form | |
| WO2008140460A1 (en) | Solid form | |
| RU2141822C1 (en) | New controlled-release granules and pharmaceutical preparations containing such granules | |
| RU2385712C2 (en) | Controlled-release formulation | |
| CA2746855C (en) | Extended-release pharmaceutical formulations | |
| US8277843B2 (en) | Programmable buoyant delivery technology | |
| US20060280795A1 (en) | Specific time-delayed burst profile delivery system | |
| KR101378973B1 (en) | Composite formulation comprising multi-unit spheroidal tablet(must) encapsulated in a hard capsule and method for preparing the same | |
| US20190133924A1 (en) | Alcohol-Resistant Formulations | |
| WO2008140461A1 (en) | Solid form | |
| RU2325163C2 (en) | Lamotrigine-based compositions of prolonged release | |
| JP2009510036A (en) | Microparticles having modified release of at least one active ingredient and oral pharmaceutical forms containing the same | |
| CA2746888A1 (en) | Misuse preventative, controlled release formulation | |
| US20100160363A1 (en) | Extended-release pharmaceutical formulations | |
| MX2012007448A (en) | Controlled release pharmaceutical composition. | |
| WO2009114773A2 (en) | Modified release formulations of anti-irritability drugs | |
| WO2011013082A1 (en) | Multi-layered, multiple unit pharmaceutical compositions | |
| CN1972674A (en) | Medicament in a multilayer form | |
| WO2006045152A1 (en) | Improved tabletting process | |
| EP1839649A1 (en) | Coated formulations for tolterodine | |
| Garg et al. | Formulation and evaluation of pulsatile drug delivery system of rosuvastatin calcium using different swelling polymers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07794919 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07794919 Country of ref document: EP Kind code of ref document: A1 |