WO2008063625A2 - Pyridine compounds and methods of their use - Google Patents
Pyridine compounds and methods of their use Download PDFInfo
- Publication number
- WO2008063625A2 WO2008063625A2 PCT/US2007/024220 US2007024220W WO2008063625A2 WO 2008063625 A2 WO2008063625 A2 WO 2008063625A2 US 2007024220 W US2007024220 W US 2007024220W WO 2008063625 A2 WO2008063625 A2 WO 2008063625A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyridin
- methyl
- morpholinomethyl
- compound according
- dimethyl
- Prior art date
Links
- 0 CC1(C)I2*(CS(C)C)CCC1C2 Chemical compound CC1(C)I2*(CS(C)C)CCC1C2 0.000 description 10
- OGJIVTCDZMXIGW-UHFFFAOYSA-N CCC(C)(C)C(Nc(cc1CN2CCOCC2)ncc1Br)=O Chemical compound CCC(C)(C)C(Nc(cc1CN2CCOCC2)ncc1Br)=O OGJIVTCDZMXIGW-UHFFFAOYSA-N 0.000 description 2
- DSMPCCHPGZQJIZ-UHFFFAOYSA-N CCC(C)(C)C(Nc1ncc(C)c(C=O)c1)=O Chemical compound CCC(C)(C)C(Nc1ncc(C)c(C=O)c1)=O DSMPCCHPGZQJIZ-UHFFFAOYSA-N 0.000 description 2
- HIJKTQACFIWLBP-UHFFFAOYSA-N O=C(c1cc(Cl)nc2c1CCC2)N1CCOCC1 Chemical compound O=C(c1cc(Cl)nc2c1CCC2)N1CCOCC1 HIJKTQACFIWLBP-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N C1NCCOC1 Chemical compound C1NCCOC1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- CNYOYKQTUDOYBB-UHFFFAOYSA-N CC(C)(C)C(Nc(nc1)cc(CN2CCOCC2)c1Br)=O Chemical compound CC(C)(C)C(Nc(nc1)cc(CN2CCOCC2)c1Br)=O CNYOYKQTUDOYBB-UHFFFAOYSA-N 0.000 description 1
- MLDCUWXMEDUWET-UHFFFAOYSA-N CC(C)(CC(F)(F)F)I Chemical compound CC(C)(CC(F)(F)F)I MLDCUWXMEDUWET-UHFFFAOYSA-N 0.000 description 1
- AMPDWXQPDUHWIU-UHFFFAOYSA-N CC(C)C(C)(C(F)(F)F)C(F)(F)F Chemical compound CC(C)C(C)(C(F)(F)F)C(F)(F)F AMPDWXQPDUHWIU-UHFFFAOYSA-N 0.000 description 1
- IAGRUEHUJYYLJX-UHFFFAOYSA-N CCC(C)(C)C(Nc(nc1)cc(CN2CCOCC2)c1-c1cccnc1)=O Chemical compound CCC(C)(C)C(Nc(nc1)cc(CN2CCOCC2)c1-c1cccnc1)=O IAGRUEHUJYYLJX-UHFFFAOYSA-N 0.000 description 1
- KXBUANIBYZETSC-UHFFFAOYSA-N CCC(C)(C)C(Nc1cc(C)cc(CBr)n1)=O Chemical compound CCC(C)(C)C(Nc1cc(C)cc(CBr)n1)=O KXBUANIBYZETSC-UHFFFAOYSA-N 0.000 description 1
- WQDVJZUNCNNRGN-UHFFFAOYSA-N CCC(C)(C)C(Nc1cc(C)cc(CN2CCOCC2)n1)=O Chemical compound CCC(C)(C)C(Nc1cc(C)cc(CN2CCOCC2)n1)=O WQDVJZUNCNNRGN-UHFFFAOYSA-N 0.000 description 1
- KCJJGMASWBRYHV-UHFFFAOYSA-N CCC(C)(C)C(Nc1nc(cccc2)c2c(N2CCOCC2)c1)=O Chemical compound CCC(C)(C)C(Nc1nc(cccc2)c2c(N2CCOCC2)c1)=O KCJJGMASWBRYHV-UHFFFAOYSA-N 0.000 description 1
- FBMWMYDLNSQEAD-UHFFFAOYSA-N CCC(C)(C)C(Nc1ncc(C)c(CO)c1)=O Chemical compound CCC(C)(C)C(Nc1ncc(C)c(CO)c1)=O FBMWMYDLNSQEAD-UHFFFAOYSA-N 0.000 description 1
- HFQIQDPEDNGZPH-UHFFFAOYSA-N CCC(c1cc(NC(C(C)(C)CC)=O)ncc1C)O Chemical compound CCC(c1cc(NC(C(C)(C)CC)=O)ncc1C)O HFQIQDPEDNGZPH-UHFFFAOYSA-N 0.000 description 1
- LIWYGYGOYLXPOG-UHFFFAOYSA-N Cc(c(CN1CCOCC1)c1)cnc1N Chemical compound Cc(c(CN1CCOCC1)c1)cnc1N LIWYGYGOYLXPOG-UHFFFAOYSA-N 0.000 description 1
- NHNDYXIBSJQYBD-UHFFFAOYSA-N Cc(nc(cc1C(OC)=O)N)c1Br Chemical compound Cc(nc(cc1C(OC)=O)N)c1Br NHNDYXIBSJQYBD-UHFFFAOYSA-N 0.000 description 1
- XXFVJWCKNWSTCY-UHFFFAOYSA-N Cc1cc(C(OC)=O)cc(N)n1 Chemical compound Cc1cc(C(OC)=O)cc(N)n1 XXFVJWCKNWSTCY-UHFFFAOYSA-N 0.000 description 1
- QNBJYUUUYZVIJP-UHFFFAOYSA-N Clc1cc(Cl)nc2c1cccc2 Chemical compound Clc1cc(Cl)nc2c1cccc2 QNBJYUUUYZVIJP-UHFFFAOYSA-N 0.000 description 1
- OEBSYGAKBYBVSJ-UHFFFAOYSA-N Clc1nc(CCC2)c2c(CN2CCOCC2)c1 Chemical compound Clc1nc(CCC2)c2c(CN2CCOCC2)c1 OEBSYGAKBYBVSJ-UHFFFAOYSA-N 0.000 description 1
- LSGAWULNENTZPN-UHFFFAOYSA-N Nc1nc(cccc2)c2c(N2CCOCC2)c1 Chemical compound Nc1nc(cccc2)c2c(N2CCOCC2)c1 LSGAWULNENTZPN-UHFFFAOYSA-N 0.000 description 1
- BYUMQKPEMYSYGR-UHFFFAOYSA-N O=C(c1cc(Cl)nc2c1CCC2)Cl Chemical compound O=C(c1cc(Cl)nc2c1CCC2)Cl BYUMQKPEMYSYGR-UHFFFAOYSA-N 0.000 description 1
- HXITXNWTGFUOAU-UHFFFAOYSA-N OB(c1ccccc1)O Chemical compound OB(c1ccccc1)O HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N OB(c1cccnc1)O Chemical compound OB(c1cccnc1)O ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- HDHQZCHIXUUSMK-UHFFFAOYSA-N OC(c(cccc1)c1N1)=CC1=O Chemical compound OC(c(cccc1)c1N1)=CC1=O HDHQZCHIXUUSMK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- This invention relates to novel pyridine compounds, pharmaceutical compositions containing such compounds, and uses thereof. More particularly, the present invention relates to novel pyridine compounds that may affect the cannabinoid receptor system and thus may be useful, inter alia, as agonists or antagonists of cannabinoid receptors.
- Cannabis sativa preparations have long been known as therapeutic agents to treat various diseases (Mechoulam, R., "Cannabinoids as Therapeutic Agents” CRC Press, Boca Raton, FIa. 1-19, 1986).
- the native active constituent, delta 9-tetrahydrocannabinol ( ⁇ 9 -THC) is prescribed today, under the generic name dronabinol, as an anti-emetic and for enhancement of appetite, mainly in AIDS patients.
- ⁇ 9 -THC delta 9-tetrahydrocannabinol
- dronabinol an anti-emetic and for enhancement of appetite, mainly in AIDS patients.
- separation between the clinically undesirable psychotropic effects and the therapeutically desirable effects on the peripheral nervous systems, the cardiovascular system, the immune and endocrine system is problematic.
- the discovery of two cannabinoid receptors, CB1 and CB2 has helped to elucidate the diverse cannabinoid effects.
- the CB1 receptor has been cloned from rat, mouse, and human tissues and exhibits 97- 99% amino acid sequence identity across species.
- the CB2 receptor exhibits 48% homology with the CB1 receptor (A.C. Howlett et al. Pharmacological Reviews 2002, 54, 161-202).
- the structures of both receptors are consistent with seven transmembrane G-protein coupled receptors.
- both receptors exert their effect by negative regulation of adenylyl cyclase activity through pertussis toxin-sensitive GTP-binding proteins. They have been shown to activate the mitogen activated protein kinase (MAPK) in certain cell types (Parolaro, D., Life Sci. 1999, 65, 637-44).
- MAPK mitogen activated protein kinase
- the CB1 receptor is expressed mainly in the central nervous systems (CNS) and to a lesser extent in other tissues including, for example, gastrointestinal tissues, immune cells, reproductive organs, heart, lung, urinary bladder, and adrenal gland.
- the CB2 receptor is expressed mostly in peripheral tissue associated with immune functions including, for example, macrophages, B cells, T cells and mast cells, as well as in peripheral nerve terminals (Perrwee, R.G., Prog. Neurobiol. 2001, 63, 569-611).
- the central distribution pattern of CB1 receptors accounts for several unwanted pharmacological properties of cannabinoids, such as impaired cognition and memory, altered control of motor function, and psychotropic and other neurobehavioral effects.
- CB1 receptors are also found on pain pathways in brain, spinal cord and at the peripheral terminals of primary sensory neurons (A. S. Rice, Curr. Opin. Investig. Drugs 2001 2(3), 399-414).
- CB1 knockout mice have been shown to be unresponsive to cannabinoids in behavioral assays providing molecular evidence that the psychotropic effects, including sedation, hallucinations, and antinociception are manifested through the activation of the CB1 receptor, present primarily in the CNS.
- Analysis of the CB2 knockout mouse has corroborated the evidence for the function of CB2 receptors in modulating the immune system.
- the CB2 receptor does not affect immune cell development and differentiation as determined by FACS analysis of cells from the spleen, lymph node and thymus from CB2 knockout mice. Further studies in these mice have shown that the immunosuppressive effects of ⁇ 9 -THC are mediated by the CB2 receptor.
- cannabinoid receptor agonists have been shown to produce potent antinociception with equivalent efficacy to morphine in animal models of acute pain, persistent inflammatory pain, and neuropathic pain. They have also been reported to induce a number of unwanted CNS side effects. Furthermore, the known cannabinoid receptor agonists are in general highly lipophilic and insoluble in water. There is thus a need for cannabinoid receptor agonists with improved properties for uses as therapeutic agents.
- CB1 cannabinoid receptor agonists produce a characteristic profile of in vivo effects in mice, including suppression of spontaneous activity, antinociception, hypothermia, and catalepsy. Measurement of these four properties, commonly referred to as the tetrad test, has played a key role in establishing the structure-activity relationship of cannabinoids and cannabimimetics acting at CB1 receptors. Catalepsy in mice is indicative of CB1 activation and predictive of cannabinoid psychoactivity. Pertwee showed a correlation between catalepsy in the ring test in mice and the previously validated dog static ataxia model (R.G. Pertwee, Br. J. Pharmacology 1972, 46, 753-763).
- catalepsy in mice is viewed as an excellent predictor of CNS effects in humans (D.R. Compton, Marijuana: An International Research Report 7, 213-218, 1987; E.W. Gill and G. Jones, Biochem. Pharmacol. 21, 2237-2248, 1972; E.W. Gill et al. Nature 228, 134-136, 1970).
- CB2 receptor-selective inverse agonists are capable of altering cellular chemotaxis mediated by either cannabinoids or chemokines, both in vivo and in vitro. They have reported that administration of these compounds can decrease allergic eosinophilia in animal models for asthma.
- the present invention is directed, in part, to novel pyridine compounds which may be modulators, agonists, and/or antagonists of cannabinoid receptors and which thus may be useful, inter alia, for the treatment of diseases or disorders which are associated with the cannabinoid receptor system.
- the present invention relates to compounds of formula I:
- A is a ring atom or a bond
- D is cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, Or -NR 2 R 3 ;
- E is N or CR 12 ;
- Y is N or CR 6 ;
- R 1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -OR x , -SR x , -NR y R z , F, Cl, or Br, or R 1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R 8 )(R 9 )] n -D; provided that: when R 1 or D independently includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7-membered ring having 0 or 1 heteroatom ring members, and when R 1 and
- R 4 is:
- each R a and each R are independently H or alkyl
- R 5 and R 5a are each independently H or alkyl
- R c is H, alkyl, or aryl
- R and R e are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
- R 6 and R 12 are each independently H, F, Cl, or alkyl, or R 6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R 8 )(R 9 )] n -D;
- R 7 is H, F, or alkyl, or R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R 7 and R 12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
- R 1 , R 6 , and R 7 are other than F;
- R 7 when R 7 is F, then at least one of R 1 and R 6 is other than F, Cl, or Br;
- R 7 and R 12 , R 1 and R 7 , R 1 and A, and R 6 and A form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
- the invention is directed to compounds according to formula Ia:
- E is N or CR 12 ;
- R 1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -OR X , -SR x , -NR y R z , F, Cl, or Br; provided that when R 1 includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members,
- R v is C 1-3 unsubstituted alkyl
- R 4 is:
- each R a and each R are independently H or alkyl;
- R 5 and R 5a are each independently H or alkyl;
- R c is H, alkyl, or aryl;
- R d and R e are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
- R 6 and R 12 are each independently H, F, Cl, or alkyl
- R 7 is H, F, or alkyl, or R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R 7 and R 12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
- R 7 and R 12 no more than one pair of R 7 and R 12 , and R 1 and R 7 form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
- the present invention is directed, in part, to pharmaceutical compositions, comprising: a pharmaceutically acceptable carrier and a compound of formula I or Ia.
- the present invention is also directed, in part, to methods of binding cannabinoid receptors in a patient in need thereof, comprising the step of administering to said patient an effective amount of a compound according to formula I or Ia.
- the present invention is generally directed to pyridine compounds, pharmaceutical compositions containing these compounds, and methods of their pharmaceutical use.
- alkyl refers to an optionally substituted, saturated, straight or branched hydrocarbon having from about 1 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), preferably with from about 1 to about 8 carbon atoms, more preferably with from about 1 to about 6 carbon atoms, yet more preferably with from about 1 to about 4 carbon atoms, still more preferably with from about 1 to about 3 carbon atoms, with 1 carbon being even more preferred.
- 1 or more of the hydrogen atoms on the alkyl group are substituted with fluorine atoms.
- Alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl, 3-methylpentyl, 2,2-dimethylbutyl, and 2,3-dimethylbutyl.
- cycloalkyl or “carbocyclic ring” each refers to an optionally substituted, mono-, di-, tri-, or other multicyclic alicyclic ring system having from about 3 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein).
- the cycloalkyl groups have from about 3 to about 8 carbon atoms, more preferably from about 3 to about 6 carbon atoms.
- Multi-ring structures may be bridged or fused ring structures, wherein the additional groups fused or bridged to the cycloalkyl ring may include optionally substituted cycloalkyl, aryl, heterocycloalkyl, or heteroaryl rings.
- exemplary cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl, adamantyl, 2-[4-isopropyl-1- methyl-7-oxa-bicyclo[2.2.1]heptanyl], and 2-[1,2,3,4-tetrahydro-naphthalenyl].
- bicycloalkyl refers to an optionally substituted, alicyclic group having two bridged rings in its structure and having from about 7 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 7 to about 15 carbon atoms being preferred.
- Exemplary bicycloalkyl-ring structures include, but are not limited to, norbornyl, bornyl, [2.2.2]-bicyclooctyl, cis-pinanyl, trans-pinanyl, camphanyl, iso-bornyl, and fenchyl.
- tricycloalkyl refers to an optionally substituted, alicyclic group having three bridged rings in its structure and having from about 7 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 7 to about 15 carbon atoms being preferred.
- Exemplary tricycloalkyl-ring structures include, but are not limited to, tricyclo[5.1.2.0 2,6 ]decane, 1,7,7-trimethyl tricyclo[2.2.1.0 2,6 ]heptane, alpha-santalol, patchouli alcohol, alpha-cedrene, and longifolene.
- cycloalkylalkyl refers to an optionally substituted ring system comprising an alkyl radical bearing one or more cycloalkyl substituents, and having from about 6 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 6 to about 10 carbon atoms being preferred, wherein alkyl and cycloalkyl are as previously defined.
- Non-limiting examples include, for example, cyclopropylmethyl, cyclobutylethyl, cyclopentylpropyl, cyclohexylmethyl, 2-cyclooctyl-1-methylethyl, 2-[4-isopropyl-1-methyl-7-oxa-bicyclo[2.2.1]heptanyl]methyl, 2-[1,2,3,4-tetrahydro-naphthalenyl]ethyl, and adamantylpropyl.
- alkylcycloalkyl refers to an optionally substituted ring system comprising a cycloalkyl group having one or more alkyl substituents, wherein cycloalkyl and alkyl are each as previously defined.
- exemplary alkylcycloalkyl groups include 2-methylcyclohexyl, 3,3-dimethylcyclopentyl, trans-2,3-dimethylcyclooctyl, and 4-methyldecahydronaphthalenyl.
- heterocycloalkyl and “heterocyclic ring” each refers to an optionally substituted ring system composed of a cycloalkyl radical wherein in at least one of the rings, one or more of the carbon atom ring members is independently replaced by a heteroatom group selected from the group consisting of O, S, N, and NH, wherein cycloalkyl is as previously defined.
- Heterocycloalkyl ring systems having a total of from about 5 to about 14 carbon atom ring members and heteroatom ring members (and all combinations and subcombinations of ranges and specific numbers of carbon and heteroatom ring members) are preferred.
- heterocyclic groups may be fused to one or more aromatic rings.
- heterocycloalkyl moieties are attached via a ring carbon atom to the rest of the molecule.
- Exemplary heterocycloalkyl groups include, but are not limited to, azepanyl, tetrahydrofuranyl, hexahydropyrimidinyl, tetrahydrothienyl, piperidinyl, pyrrolidinyl, isoxazolidinyl, isothiazolidinyl, pyrazolidinyl, oxazolidinyl, thiazolidinyl, piperazinyl, 2-oxo-morpholinyl, morpholinyl, 2-oxo-piperidinyl, piperadinyl, decahydroquinolyl, octahydrochromenyl, octahydro-cyclopenta[c]pyranyl, 1,2,3 ,4,-t
- two moieties attached to a heteroatom may be taken together to form a heterocycloalkyl ring, such as when R and R , taken together with the nitrogen atom to which they are attached, form a heterocycloalkyl ring.
- the resultant ring when a moiety containing one ring replacement atom replaces a ring carbon atom, the resultant ring, after replacement of a ring atom by the moiety, will contain the same number of ring atoms as the ring before ring atom replacement.
- the resultant ring after replacement will contain one more ring atom than the ring prior to replacement by the moiety.
- the resultant ring is a 7-membered ring containing 2 ring nitrogen atoms and the carbon of a carbonyl group in addition to 4 other carbon ring atoms (CH 2 groups) from the original piperidine ring.
- heterocycloalkylalkyl refers to an optionally substituted ring system composed of an alkyl radical having one or more heterocycloalkyl substituents, wherein heterocycloalkyl and alkyl are as previously defined.
- the alkyl moieties of the heterocycloalkylalkyl groups have from about 1 to about 3 carbon atoms.
- heterocycloalkyl groups include, but are not limited to, azepanylmethyl, tetrahydrofuranylethyl, hexahydropyrimidinylisobutyl, tetrahydrothienylpropyl, piperidinyl-2,2- dimethylethyl, pyrrolidinylmethyl , isoxazolidinylethyl, isothiazolidinylpropyl, pyrazolidinylmethyl, oxazolidinylbutyl, thiazolidinylisopropyl, piperazinylmethyl, 2-oxo-morpholinylmethyl, morpholinylethyl, 2-oxo-piperidinylethyl, piperadinylmethyl, decahydroquinolylethyl, octahydrochromenylpropyl, octahydro-cyclopenta[c]pyranylbutyl, 1,2,3,4,-te
- alkenyl refers to an optionally substituted alkyl group having from about 2 to about 10 carbon atoms and one or more double bonds (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), wherein alkyl is as previously defined.
- alkynyl refers to an optionally substituted alkyl group having from about 2 to about 10 carbon atoms and one or more triple bonds (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), wherein alkyl is as previously defined.
- aryl refers to an optionally substituted, mono-, di-, tri-, or other multicyclic aromatic ring system having from about 6 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), preferably with from about 6 to about 10 carbons, with about 6 carbon atoms being preferred.
- Non-limiting examples include, for example, phenyl, naphthyl, anthracenyl, and phenanthrenyl.
- aryl is phenyl, optionally substituted, more preferably substituted with at least one halo or haloalkyl group.
- aralkyl refers to an optionally substituted ring system comprising an alkyl radical bearing an aryl substituent and having from about 7 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), preferably with from about 7 to about 11 carbon atoms, with about 7 carbon atoms being preferred.
- Non-limiting examples include, for example, benzyl, diphenylmethyl, triphenylmethyl, phenylethyl, and diphenylethyl.
- alkylaralkyl refers to an optionally substituted ring system comprising an alkyl radical bearing an aralkyl substituent and having from about 8 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 8 to about 12 carbon atoms being preferred, wherein alkyl and aralkyl are as previously defined.
- Non-limiting examples include, for example, to lylm ethyl, bis(isopropylphenyl)methyl, 1-tolyl-1-ethylphenylmethyl, tert-butylphenylethyl, and ort ⁇ o-methyl-jp ⁇ ra-butylphenylethyl.
- alkoxyl refers to an optionally substituted alkyl-O- group wherein alkyl is as previously defined.
- the alkyl moieties of the alkoxy groups have from about 1 to about 4 carbon atoms.
- Exemplary alkoxy groups include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, and heptoxy.
- aryloxyl refers to an optionally substituted aryl-O- group wherein aryl is as previously defined.
- Exemplary aryloxy groups include, but are not limited to, phenoxy and naphthoxy.
- aralkoxyl refers to an optionally substituted aralkyl-O- group wherein aralkyl is as previously defined.
- exemplary aralkoxy groups include, but are not limited to, benzyloxy, 1-phenylethoxy, 2-phenylethoxy, and 3-naphthytheptoxy.
- 1,1-dioxo-thio refers to a diradical having the structure:
- a 1 , 1 -dioxo-thio is a ring member of a heterocyclic ring.
- exemplary ring member 1,1-dioxo-groups such as 1,1-dioxo-thiirane, 1,1-dioxo-thietane, 1,1-dioxo- tetrahydrothiophene, 1,1-dioxo-thiopyran, 1,1-dioxo-thiepane, and the like, more preferably 1,1- dioxo-thiopyran.
- one or more ring carbon atoms of a 1,1-dioxo-thio ring is optionally replaced by -O-, -S-, or -N(R 9a )-.
- Exemplary rings include 1,1-dioxo- thiomorpholine and [1,4]oxathiane 4,4-dioxide.
- halo refers to a fluoro, chloro, bromo, or iodo moiety, with fluoro, chloro, or bromo moieties being preferred. In certain embodiments, fluoro or chloro moieties are more preferred.
- heteroaryl refers to an optionally substituted aryl ring system wherein in at least one of the rings, one or more of the carbon atom ring members is independently replaced by a heteroatom group selected from the group consisting of S, O, N, and NH, wherein aryl is as previously defined.
- Heteroaryl groups having a total of from about 5 to about 14 carbon atom ring members and heteroatom ring members(and all combinations and subcombinations of ranges and specific numbers of carbon and heteroatom ring members) are preferred.
- heteroaryl groups include, but are not limited to, pyrryl, furyl, pyridyl, pyridine-N-oxide, 1,2,4-thiadiazolyl, pyrimidyl, thienyl, isothiazolyl, imidazolyl, tetrazolyl, pyrazinyl, pyrimidyl, quinolyl, isoquinolyl, thiophenyl, benzothienyl, isobenzofuryl, pyrazolyl, indolyl, purinyl, carbazolyl, benzimidazolyl, and isoxazolyl.
- Heteroaryl may be attached via a carbon or a heteroatom to the rest of the molecule.
- heteroarylalkyl refers to an optionally substituted ring system comprising an alkyl radical bearing a heteroaryl substituent, having from about 2 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 6 to about 25 carbon atoms being preferred.
- Non- limiting examples include 2-(1H-pyrrol-3-yl)ethyl, 3-pyridylmethyl, 5-(2H-tetrazolyl)methyl, and 3 -(pyrimidin-2-yl)-2-methylcyclopentanyl.
- spiroalkyl refers to an optionally substituted alkylene diradical, both ends of which are bonded to the same carbon atom of the parent group to form a spirocyclic group.
- the spirocyclic group as herein defined, has 3 to 20 ring atoms, preferably with 3 to 10 ring atoms.
- Exemplary spiroalkyl groups taken together with its parent group include, but are not limited to, 1-(1-methyl-cyclopropyl)-propan-2-one, 2-(1-phenoxy- cyclopropyl)-ethylamine, and 1-methyl-spiro[4.7]dodecane.
- substituted chemical moieties include one or more substituents that replace hydrogen.
- each moiety R" can be, independently, any of H, alkyl, cycloalkyl, alkenyl, aryl, aralkyl, heteroaryl, or heterocycloalkyl, or when (R"(R")) is attached to a nitrogen atom, R" and R" can be taken together with the nitrogen atom to which they are attached to form a 4- to 8-membered nitrogen heterocycle, wherein the heterocycloalkyl ring is optionally interrupted by one or more additional -O-, -S-, -SO, -SO 2 -, -NH-, -N(alkyl)-, or -N(aryl)- groups, for example,
- chemical moieties are substituted by at least one optional substituent, such as those provided hereinabove.
- the optional substituents are not further substituted.
- R 1 is an alkyl moiety, it is optionally substituted, based on the definiton of "alkyl" as set forth herein.
- R 1 is alkyl substituted with optional aryl, the optional aryl substituent is not further substituted.
- 2-(alpha-naphthyl)ethyl is within the scope of optionally substituted alkyl.
- 2-(3-chlorophenyl)ethyl (wherein ethyl is the alkyl moiety and 3-chlorophenyl is the optional substituent) is not within the scope of optionally substituted alkyl because the optional aryl substiuent cannot be further substituted by a further chemical group.
- cannabinoid refers to any one of a group of naturally occurring compounds of related structure that may be isolable from Cannabis sativa, more commonly known as marijuana, and structurally modified derivatives thereof.
- Cannabinoids include for example, compounds such as ⁇ 9 -tetrahydrocannabinol, ⁇ 8 -tetrahydrocannabinol, cannabichromene, cannabicyclol, cannabidiol, cannabielsoin, cannabigerol, cannabinol, cannabitriol, nabilone, and nantradol, and numerous structural variants.
- cannabinoids are lipophilic and have low solubility in water.
- cannabinoids refers to any of a group of endogenous or exogenous receptor ligands that bind one or more of the receptors bound by cannabinoids and mimic one or more behaviors of cannabinoids while so bound.
- Examples of endogenous cannabimimetics (also referred to as “endocannabinoids”) produced in mammalian tissues include, for example, arachidonoylethanolamide (anandamide), 2-arachidonoyl glycerol, 1(3)- arachidonoyl glycerol, and palmitoylethanolamide.
- exogenous cannabimimetics examples include, for example WIN 55,212-2, CP 55,940, HU-210, and the like.
- exogenous cannbimimetics may be found in publications such as R.B. Pertwee, "Pharmacology of Cannabinoid Receptor Ligands", Current Medicinal Chemistry, 1999, 6, 635-664, and A.C. Howlett, et al. "International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors", Pharmacological Reviews, 2002, 54(2), 161-202, the disclosures of which are each hereby incorporated herein by reference in their entireties.
- the term "antagonist” refers to a compound that binds to a receptor to form a complex that preferably does not elicit any response, in the same manner as an unoccupied receptor, and does not alter the equilibrium between inactive and active receptor.
- agonist refers to a ligand that produces a conformational change in the receptor and alters the equilibrium of the receptor's active and inactive states, which in turn induces a series of events, resulting in a measurable biological response.
- Agonists include, for example, conventional agonists, which exhibit positive receptor activity, and inverse agonists, which exhibit a negative intrinsic activity.
- prodrug refers to compounds that may serve to maximize the amount of active species that reaches the desired site of reaction that are themselves typically inactive or minimally active for the activity desired, but through biotransformation are converted into biologically active metabolites.
- stereoisomers refers to compounds that have identical chemical constitution, but differ as regards the arrangement of the atoms or groups in space.
- partial stereoisomers refers to stereoisomers having two or more chiral centers wherein at least one of the chiral centers has defined stereochemistry (i.e., R or S) and at least one has undefined stereochemistry (i.e., R or S).
- R or S defined stereochemistry
- R or S undefined stereochemistry
- stereoisomer has three chiral centers and the stereochemical configuration of the first center is defined as having "S" stereochemistry
- the term "or partial stereoisomer thereof refers to stereoisomers having SRR, SRS, SSR, or SSS configurations at the three chiral centers, and mixtures thereof.
- Asymmetric carbon atoms may be introduced into the molecule depending on the structure of the moiety R 4 when R a and R b are non-identical or when R c , R d , and R e are non- identical.
- R 4 when R a is hydrogen and R b is other than H, the carbon atom to which R a is attached is asymmetric.
- asymmetric centers are contemplated in the present invention.
- Asymmetric centers are, by convention, present in R 4 moieties structure such as those shown below at the ring carbon atoms identified with an asterisk (*). As such, these classes of compounds can exist as
- N-oxide refers to compounds wherein the basic nitrogen atom of either a heteroaromatic ring or tertiary amine is oxidized to give a quaternary nitrogen bearing a positive formal charge and an attached oxygen atom bearing a negative formal charge.
- hydrate refers to a compound of the present invention which is associated with water in the molecular form, i.e., in which the H-OH bond is not split, and may be represented, for example, by the formula R ⁇ 2 O, where R is a compound of the invention.
- R is a compound of the invention.
- a given compound may form more than one hydrate including, for example, monohydrates (R . H 2 O) or polyhydrates (R . nH 2 O wherein n is an integer > 1) including, for example,
- solvate refers to a compound of the present invention which is associated with solvent in the molecular form, i.e., in which the solvent is coordinatively bound, and may be represented, for example, by the formula Resolvent), where R is a compound of the invention.
- a given compound may form more than one solvate including, for example, monosolvates (R . (solvent)) or polysolvates (R . n(solvent)) wherein n is an integer > 1) including,
- Solvents herein include mixed solvents, for example, methanol/water, and as such, the solvates may incorporate one or more solvents within the solvate.
- the term "acid hydrate” refers to a complex that may be formed through association of a compound having one or more base moieties with at least one compound having one or more acid moieties or through association of a compound having one or more acid moieties with at least one compound having one or more base moieties, said complex being further associated with water molecules so as to form a hydrate, wherein said hydrate is as previously defined and R represents the complex herein described above.
- the term "pharmaceutically acceptable salts” refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like
- organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic,
- physiologically acceptable salts are prepared by methods known in the art, e.g., by dissolving the free amine bases with an excess of the acid in aqueous alcohol, or neutralizing a free carboxylic acid with an alkali metal base such as a hydroxide, or with an amine.
- the term "effective amount” refers to an amount of a compound as described herein that may be therapeutically effective to inhibit, prevent or treat the symptoms of particular disease, disorder or side effect.
- diseases, disorders and side effects include, but are not limited to, those pathological conditions associated with the binding of cannabinoid receptors (for example, in connection with the treatment and/or prevention of pain), wherein the treatment or prevention comprises, for example, agonizing the activity thereof by contacting cells, tissues or receptors with compounds of the present invention.
- the term "effective amount,” when used in connection with cannabinoids, for example, for the treatment of pain refers to the treatment and/or prevention of the painful condition.
- the term "effective amount”, when used in connection with the present cannabinoid receptor agonist compounds refers to the treatment, reduction and/or prevention of side effects typically associated with cannabinoids including, for example, such side effects as those hereinabove mentioned.
- the term "pharmaceutically acceptable” refers to those compounds, materials, compositions, and/or dosage forms that are, within the scope of sound medical judgment, suitable for contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problems or complications commensurate with a reasonable benefit/risk ratio.
- the term “in combination with”, “combination therapy” and “combination products” refer, in certain embodiments, to the concurrent administration to a patient of cannabinoids and the compounds of the Formula I, II, III, or IV.
- each component When administered in combination, each component may be administered at the same time or sequentially in any order at different points in time. Thus, each component may be administered separately but sufficiently closely in time so as to provide the desired therapeutic effect.
- the term “dosage unit” refers to physically discrete units suited as unitary dosages for the particular individual to be treated. Each unit may contain a predetermined quantity of active compound(s) calculated to produce the desired therapeutic effect(s) in association with the required pharmaceutical carrier.
- the specification for the dosage unit forms of the invention may be dictated by (a) the unique characteristics of the active compound(s) and the particular therapeutic effect(s) to be achieved, and (b) the limitations inherent in the art of compounding such active compound(s).
- treatment includes preventative (e.g., prophylactic), curative or palliative treatment and “treating” as used herein also includes preventative, curative and palliative treatment.
- pain refers to the perception or condition of unpleasant sensory or emotional experience, which may or may not be associated with actual or potential tissue damage or described in terms of such damage.
- Pain includes, but is not limited to, two broad categories of pain: acute and chronic pain (Buschmann, H.; Christoph, T; Friderichs, E.; Maul, C; Sundermann, B; eds.; Analgesics, Wiley- VCH, Verlag GMbH & Co. KgaA, Weinheim; 2002; Jain, K. K. "A Guide to Drug Evaluation for Chronic Pain”; Emerging Drugs, 5(2), 241-257(2000)).
- Non-limiting examples of pain include nociceptive pain, inflammatory pain, visceral pain, somatic pain, neuropathic pain, AIDS pain, cancer pain, phantom pain, and psychogenic pain, and pain resulting from hyperalgesia, pain caused by rheumatoid arthritis, migraine, allodynia, and the like.
- the term "patient” refers to animals, including mammals, preferably humans.
- side effect refers to a consequence other than the one(s) for which an agent or measure is used, as the adverse effects produced by a drug, especially on a tissue or organ system other then the one sought to be benefited by its administration.
- the term “side effect” may refer to such conditions as, for example, psychotropic effects, such as confusion, anxiety, panic, distortion of perception, pretendizing, sedation, inner unrest, irritability and insomnia, sweating, rhinorrhoea, loose stools, hiccups, dry mouth, tachycardia, ataxia, dizziness, orthostatic hypotension, and anorexia.
- any variable occurs more than one time in any constituent or in any formula, its definition in each occurrence is independent of its definition at every other occurrence. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- the present invention is directed, in part, to a new class of cannabinoid receptor modulator compounds, preferably pyridine compounds, which may be highly useful in connection with the binding of cannabinoid receptors.
- Compounds binding cannabinoid receptors may agonize and/or antagonize the receptors.
- a cannabimimetic compound or ligand agonizes one or more cannabinoid receptors the resultant binding is believed to trigger an event or series of events in the cell that results in a change in the cell's activity, its gene regulation, or the signals that it sends to neighboring cells, similar to that of a cannabinoid.
- compounds of the invention may serve to prevent or treat diseases or disorders in which cannabinoid receptors are implicated.
- a cannabimimetic compound or ligand antagonizes one or more cannabinoid receptors
- the resultant binding typically occurs comparatively to a greater extent relative to that of the cannabinoid, but does not trigger one or more of the events of signal transduction.
- Compounds with these properties are highly useful, for example, in connection with the study of functions of cannabinoid receptors, which may result, for example, in the development of new cannabimimetic agonist compounds, such as those, for example, reported in Rinaldi-Carmona, M. et al., Journal of Pharmacology and Experimental Therapeutics, 1998, 284(2), 644-650, the disclosure of which is hereby incorporated herein by reference, in its entirety.
- the present invention provides compounds of formula I: wherein:
- A is a ring atom or a bond
- D is cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, Or -NR 2 R 3 ;
- E is N or CR 12 ;
- Y is N or CR 6 ;
- R 1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -OR x , -SR X , -NR y R z , F, Cl, or Br, or R 1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R 8 )(R 9 )] n -D; provided that: when R 1 or D independently includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7-membered ring having 0 or 1 heteroatom ring members, and when R 1 and
- R 4 is:
- each R a and each R D are independently H or alkyl
- R 5 and R 5a are each independently H or alkyl
- R c is H, alkyl, or aryl
- R d and R e are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
- R 6 and R 12 are each independently H, F, Cl, or alkyl, or R 6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R 8 )(R 9 )] n -D;
- R 7 is H, F, or alkyl, or R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R 7 and R 12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
- R 1 , R 6 , and R 7 are other than F;
- R 7 when R 7 is F, then at least one of R 1 and R 6 is other than F, Cl, or Br;
- R 7 and R 12 , R 1 and R 7 , R 1 and A, and R 6 and A form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
- the present invention provides compounds of formula Ia:
- E is N or CR 1 1 2.
- Y is N or CR 6 ;
- R 1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -OR X , -SR X , -NR y R z , F, Cl, or Br; provided that when R 1 includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members,
- R v is C 1-3 unsubstituted alkyl
- R 4 is:
- each R a and each R are independently H or alkyl
- R 5 and R 5a are each independently H or alkyl
- R c is H, alkyl, or aryl
- R and R e are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
- R 6 and R 12 are each independently H, F, Cl, or alkyl
- R 7 is H, F, or alkyl, or R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R 7 and R 12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
- R 7 and R 12 no more than one pair of R 7 and R 12 , and R 1 and R 7 form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
- the compound of formula I or Ia has the structure of formula II:
- R 1 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , A, D, and n are as defined above.
- the compound of formula I or Ia has the structure of formula III: wherein R 1 , R 4 , R 7 , R 8 , R 9 , A, D, and n are as defined above.
- the compound of formula I has the structure of formula IV:
- R 4 , R 7 , R 8 , R 9 , D, E, Y, Z, and n are as defined above.
- A is a bond.
- D is -NR 2 R 3 .
- D is heterocycloalkyl containing at least one nitrogen atom or dioxo-thio group; more preferably heterocycloalkyl containing at least one nitrogen atom.
- D is preferably morpholinyl, piperidinyl, or 1,1-dioxo-thiomorpholinyl, each optionally substituted.
- D is N(H)-aralkyl or -N(CH 3 )-aralkyl.
- E is N.
- R 1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, hetero aralkyl, -OR X , -SR X , -NR y R z , F, Cl, or Br, more preferably H, alkyl, cycloalkyl, aryl, heteroaryl, -OR X , or Br, with alkyl, -OR X , or Br being even more preferred.
- R 1 is H.
- R 1 is -OR X , more preferably alkoxy. In still other preferred embodiments, R 1 is alkyl. In certain other preferred embodiments, when R 1 includes a cycloalkyl, aryl, heterocycloalkyl or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members.
- the term "included ring moiety" refers to when any element independently, for example, R 1 , is a group having a ring moiety within the group.
- R 1 is the group cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl.
- These groups respectively comprise the ring moiety cycloalkyl, cycloalkyl, aryl, aryl, heterocycloalkyl, heterocycloalkyl, heteroaryl, or heteroaryl.
- groups such as aralkyl, heterocycloalkylalkyl, and heteroaralkyl which are alkyl moieties substituted with an aryl, heterocycloalkyl, and heteroaryl ring moieties respectively, have an included ring moiety.
- R 1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 7-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R )(R )] n -D, and may be further optionally substituted.
- R 1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group
- the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic ring system, wherein the monocyclic ring group is substituted with -[C(R 8 )(R 9 )] n -D.
- the -[C(R 8 )(R 9 )] n -D moiety is attached to the monocyclic ring carbon atom which is both adjacent to, and closer in proximity to the Y moiety in the core pyridine ring in the compound of formula I, II, or III.
- R 1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 6-membered aryl ring
- the resulting fused bicyclic ring system which is formed will be a quinoline group or isoquinoline group, depending upon which of E and Y is N.
- Y is N
- the resulting fused bicyclic ring system is a substituted quinoline group.
- the resulting fused bicyclic ring system is a substituted isoquinoline group.
- the formed monocyclic ring group has 0 or 1 heteroatom ring members.
- R x , R y , and R z are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl.
- R x is alkyl.
- R 2 and R 3 are each independently alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl, more preferably when at least one of R 2 and R 3 is aralkyl or cycloalkyl; still more preferably when at least one of R 2 and R 3 is aralkyl, with when one of R 2 and R 3 is aralkyl and the other is alkyl being even more preferred.
- R 3 is H or alkyl.
- R 2 and R 3 are taken together with the nitrogen atom to which they are attached to form a heterocycloalkyl ring, the ring is fused to a C 6 -aryl ring. In certain other embodiments wherein R 2 and R 3 are taken together with the nitrogen atom to which they are attached to form a heterocycloalkyl ring, the heterocycloalkyl ring is substituted with at least one hydroxy, alkyl, dialkylamino, halo, heteroarylcarbonyl, or alkylcarbonyl group. In still other alternative embodiments, R 2 and R 3 are each independently alkyl.
- R 4 is :
- R 4 is:
- R 5 is H. In other preferred embodiments of the compounds of formula I or IV, R 5a is H.
- each R a and each R b are independently H or alkyl.
- each R a is H.
- each R b is H.
- R a and R b are each H.
- R 6 is H.
- R 6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 8-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R 8 )(R 9 )] n -D, and may be further optionally substituted .
- R 6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group
- the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic bicyclic ring system, wherein the monocyclic ring group is substituted with -[C(R 8 )(R 9 )] n -D.
- the -[C(R 8 )(R 9 )] n -D moiety is attached to the monocyclic ring carbon atom which is either on a ring carbon alpha or beta to the core pyridine ring, and closer in proximity to the R 1 moiety in the compound of formula I or II.
- the resulting fused bicyclic ring system which is formed will be an isoquinoline group.
- the formed monocyclic ring group has 0 or 1 heteroatom ring members.
- R c is H, alkyl, or aryl, preferably H or alkyl, with alkyl being more preferred. In some preferred embodiments where R c is substituted alkyl, R c is preferably alkoxy substituted alkyl. In other preferred embodiments where R c is substituted aryl, R c is preferably alkoxy substituted aryl.
- R d and R e are each independently H or alkyl, more preferably alkyl.
- R c , R d , and R e are each independently alkyl.
- R d or R e is substituted alkyl, it is more preferably alkoxy substituted alkyl.
- R d and R e taken together with the carbon atom to which they are attached form a carbocyclic ring, preferably a monocyclic carbocyclic ring.
- the carbocyclic ring is further substituted, preferably with at least one alkyl or alkoxy group, or any combination thereof.
- R d and R e taken together with the carbon atom to which they are attached, form a 3- to 12-membered carbocyclic ring, wherein the carbocyclic ring is substituted with 0-5 groups each independently selected from d.C 4 alkyl and C 1 .C 4 alkoxyl. More preferably, the carbocyclic ring is bicycloalkyl or tricycloalkyl, still more preferably bicycloalkyl; with bicycloalkyl ring is substituted with 1-3 alkyl, preferably groups.
- R 7 is H or alkyl. In other preferred embodiments, R 7 is F.
- R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 4- to 7- membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 7-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R 8 )(R 9 )] n -D, and may be further optionally substituted .
- R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group
- the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic ring system.
- R 1 and R 7 taken together with the atoms through which they are connected form a monocyclic 6-membered aryl ring
- the resulting fused bicyclic ring system which is formed will be a quinoline group or isoquinoline group, depending upon which of E and Y is N.
- the resulting fused bicyclic ring system is an isoquinoline group.
- E is N
- the resulting fused bicyclic ring system is a quinoline group.
- R 1 and R 7 taken together with the atoms through which they are connected form a ring
- the formed monocyclic ring group has 0 or 1 heteroatom ring members.
- R 1 and R 7 taken together with the atoms through which they are attached form an aryl ring, even more preferably a C 6 aryl ring.
- R 7 and R 12 taken together with the atoms through which they are connected form a monocyclic 4- to 8- membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 8-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R 8 )(R 9 )] n -D, and may be further optionally substituted .
- R 7 and R 12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group
- the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic ring system.
- the resulting fused bicyclic ring system which is formed will be an isoquinoline group.
- the formed monocyclic ring group has 0 or 1 heteroatom ring members.
- R and R are each H. Alternatively, they are each independently alkyl.
- R 12 is H.
- n and r are each independently 0, 1, 2, or 3, preferably 0, 1, or 2, more preferably 0 or 1. In certain even more preferred embodiments r is 0. In other even more preferred embodiments, n is 1.
- the compound of formula I is selected from the group consisting of:
- the compound is selected from the group consisting of:
- the compound of formula I is selected from the group consisting of:
- the compound of formula I is selected from the group consisting of:
- the compound of formula I is selected from the group consisting of: N-((l/?,2i?,4S)-bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H- cyclopenta[b]-pyridin-2-amine;
- the compound of formula I is selected from the group consisting of:
- the compound of formula Ia is selected from the group consisting of: N-[4-(l , 1 -Dioxo- 1 -lambda *6*-thiomorpholin-4-yl-methyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyr amide;
- the compound of formula Ia is selected from the group consisting of:
- prodrug is intended to include any covalently bonded carriers which release the active parent drug, for example, as according to Formula I, Ia, II, III, or IV, or other formulas or compounds employed in the methods of the present invention in vivo when such prodrug is administered to a mammalian subject. Since prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc.) the compounds employed in the present methods may, if desired, be delivered in prodrug form. Thus, the present invention contemplates methods of delivering prodrugs.
- Prodrugs of the compounds employed in the present invention may be prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
- prodrugs include, for example, compounds described herein in which a hydroxy, amino, or carboxy group is bonded to any group that, when the prodrug is administered to a mammalian subject, cleaves to form a free hydroxyl, free amino, or carboxylic acid, respectively.
- Examples include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups; and alkyl, carbocyclic, aryl, and alkylaryl esters such as methyl, ethyl, propyl, iso-propyl, butyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, phenyl, benzyl, and phenethyl esters, and the like.
- the compounds employed in the methods of the present invention may be prepared in a number of ways well known to those skilled in the art. The compounds can be synthesized, for example, by the methods described below, or variations thereon as appreciated by the skilled artisan. All processes disclosed in association with the present invention are contemplated to be practiced on any scale, including milligram, gram, multigram, kilogram, multikilogram or commercial industrial scale.
- compounds employed in the present methods may contain one or more asymmetrically substituted carbon atoms, and may be isolated in optically active or racemic forms.
- optically active or racemic forms all chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated.
- mixtures of stereoisomers may be separated by standard techniques including, but not limited to, resolution of racemic forms, normal, reverse-phase, and chiral chromatography, preferential salt formation, recrystallization, and the like, or by chiral synthesis either from chiral starting materials or by deliberate synthesis of target chiral centers.
- protecting groups present may contain protecting groups during the course of synthesis.
- Protecting groups are known per se as chemical functional groups that can be selectively appended to and removed from functionalities, such as hydroxyl groups and carboxy groups. These groups are present in a chemical compound to render such functionality inert to chemical reaction conditions to which the compound is exposed.
- Any of a variety of protecting groups may be employed with the present invention.
- Preferred protecting groups include the benzyloxycarbonyl group and the tert-butyloxycarbonyl groups.
- Preferred hydroxyl protecting groups include the benzyl and the tertiary-butyldimethylsilyl groups.
- Other preferred protecting groups that may be employed in accordance with the present invention may be described in Greene, T.W.
- the compounds of the present invention are preferably combined with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice as described, for example, in Remington 's Pharmaceutical Sciences (Mack Pub. Co., Easton, PA, 1980), the disclosure of which is hereby incorporated herein by reference, in its entirety.
- a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice as described, for example, in Remington 's Pharmaceutical Sciences (Mack Pub. Co., Easton, PA, 1980), the disclosure of which is hereby incorporated herein by reference, in its entirety.
- the compounds of the present invention may be administered as the pure chemicals, it is preferable to present the active ingredient as a pharmaceutical composition.
- the invention thus further provides pharmaceutical compositions comprising one or more of the cannabinoid receptor modulator compounds of the present invention, for example, compounds of Formula I, Ia, II, III, or IV, together with one or more pharmaceutically acceptable carriers therefore and, optionally, other therapeutic and/or prophylactic ingredients.
- the carrier(s) must be acceptable in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
- compositions and methods of the invention may further comprise at least one cannabinoid.
- cannabinoids are available that may be suitable for use in the present methods and compositions.
- the cannabinoid provide the desired effect (for example, pain alleviation), and be capable of being incorporated into the present combination products and methods (discussed in detail below).
- the present methods and compositions may involve a cannabinoid that is selected from ⁇ -tetrahydrocannabinol and cannabidiol, and mixtures thereof, more preferably, ⁇ 9 -tetrahydrocannabinol.
- compositions and methods of the invention may further comprise at least one opioid.
- opioids are available that may be suitable for use in the present methods and compositions. Generally speaking, it is only necessary that the opioid provide the desired effect (for example, pain alleviation), and be capable of being incorporated into the present combination products and methods (discussed in detail below).
- the present methods and compositions may involve an opioid that is selected from alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil and/or tramadol.
- an opioid that is selected from alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentan
- the opioid is selected from morphine, codeine, oxycodone, hydrocodone, dihydrocodeine, propoxyphene, fentanyl, tramadol, and mixtures thereof.
- the opioid component of the present methods and compositions may further include one or more other active ingredients that may be conventionally employed in analgesic and/or cough-cold-antitussive combination products.
- Such conventional ingredients include, for example, aspirin, acetaminophen, phenylpropanolamine, phenylephrine, chlorpheniramine, caffeine, and/or guaifenesin.
- Typical or conventional ingredients that may be included in the opioid component are described, for example, in the Physicians ' Desk Reference, 1999, the disclosure of which is hereby incorporated herein by reference, in its entirety.
- the opioid component may further include one or more compounds that may be designed to enhance the analgesic potency of the opioid and/or to reduce analgesic tolerance development.
- compounds include, for example, dextromethorphan or other NMDA antagonists (Mao, M. J., et al, Pain, 1996, 67, 361), L-364,718 and other CCK antagonists (Dourish, C.T., et al, Eur. J. Pharmacol, 1988, 147, 469), NOS inhibitors (Bhargava, H. N., et al., Neuropeptides, 1996, 30, 219), PKC inhibitors (Bilsky, E. J., et al, J. Pharmacol.
- compositions of the invention may further comprise at least one analgesic, such as for example, COX2 inhibitors, aspirin, acetaminophen, ibuprophen, naproxen, and the like, and mixtures thereof.
- analgesic such as for example, COX2 inhibitors, aspirin, acetaminophen, ibuprophen, naproxen, and the like, and mixtures thereof.
- COX2 inhibitors such as for example, COX2 inhibitors, aspirin, acetaminophen, ibuprophen, naproxen, and the like, and mixtures thereof.
- analgesic such as for example, COX2 inhibitors, aspirin, acetaminophen, ibuprophen, naproxen, and the like, and mixtures thereof.
- the analgesic provide the desired effect (for example, pain alleviation), and be capable of being incorporated into the present combination products and methods (discussed in detail below).
- compositions of the invention may further comprise at least one therapeutic agent selected from the group consisting of anti-seizure agents, such as, for example, carbamazepine, gabapentin, lamotrigine, and phenytoin, anti-depressants such as, for example, amitryptiline, NMDA receptor antagonists, ion channel antagonists, nicotinic receptor agonists, and anti -Parkinson's agents, such as, for example, deprenyl, amantadine, levodopa, and carbidopa.
- anti-seizure agents such as, for example, carbamazepine, gabapentin, lamotrigine, and phenytoin
- anti-depressants such as, for example, amitryptiline, NMDA receptor antagonists, ion channel antagonists, nicotinic receptor agonists
- anti -Parkinson's agents such as, for example, deprenyl
- the anti seizure agent, anti-depressant, NMDA receptor antagonist, ion channel antagonist, nicotinic receptor agonist, or antiParkinson's agent provide the desired effect (for example, inhibition of seizures, alleviation of depression, and the like), and be capable of being incorporated into the present combination products and methods (discussed in detail below).
- the compounds of the invention may be administered in an effective amount by any of the conventional techniques well-established in the medical field.
- the compounds employed in the methods of the present invention including, for example, the compounds of Formula I, Ia, II, III, or IV, may be administered by any means that results in the contact of the active agents with the agents' site or site(s)of action in the body of a patient.
- the compounds may be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. For example, they may be administered as the sole active agents in a pharmaceutical composition, or they can be used in combination with other therapeutically active ingredients.
- Compounds of the present invention can be administered to a mammalian host in a variety of forms adapted to the chosen route of administration, e.g., orally or parenterally.
- Parenteral administration in this respect includes administration by the following routes: intravenous, intramuscular, subcutaneous, intraocular, intrasynovial, transepithelial including transdermal, ophthalmic, sublingual and buccal; topically including ophthalmic, dermal, ocular, rectal and nasal inhalation via insufflation, aerosol and rectal systemic.
- the active compound may be orally administered, for example, with an inert diluent or with an assimilable edible carrier, or it may be enclosed in hard or soft shell gelatin capsules, or it may be compressed into tablets, or it may be incorporated directly with the food of the diet.
- the active compound may be incorporated with excipient and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
- the amount of active compound(s) in such therapeutically useful compositions is preferably such that a suitable dosage will be obtained.
- Preferred compositions or preparations according to the present invention may be prepared so that an oral dosage unit form contains from about 0.1 to about 1000 mg of active compound.
- the tablets, troches, pills, capsules and the like may also contain one or more of the following: a binder, such as gum tragacanth, acacia, corn starch or gelatin; an excipient, such as dicalcium phosphate; a disintegrating agent, such as corn starch, potato starch, alginic acid and the like; a lubricant, such as magnesium stearate; a sweetening agent such as sucrose, lactose or saccharin; or a flavoring agent, such as peppermint, oil of wintergreen or cherry flavoring.
- a binder such as gum tragacanth, acacia, corn starch or gelatin
- an excipient such as dicalcium phosphate
- a disintegrating agent such as corn starch, potato starch, alginic acid and the like
- a lubricant such as magnesium stearate
- a sweetening agent such as sucrose, lactose or saccharin
- a flavoring agent such
- any material used in preparing any dosage unit form is preferably pharmaceutically pure and substantially non-toxic in the amounts employed.
- the active compound may be incorporated into sustained-release preparations and formulations.
- the active compound may also be administered parenterally or intraperitoneally.
- Solutions of the active compounds as free bases or pharmacologically acceptable salts can be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose.
- a dispersion can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. Under ordinary conditions of storage and use, these preparations may contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include, for example, sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form is preferably sterile and fluid to provide easy syringability. It is preferably stable under the conditions of manufacture and storage and is preferably preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier may be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol and the like), suitable mixtures thereof, and vegetable oils.
- the proper fluidity can be maintained, for example, by the use of a coating, such as lecithin, by the maintenance of the required particle size in the case of a dispersion, and by the use of surfactants.
- a coating such as lecithin
- surfactants for example, sodium sulfate, sodium sulfate, sodium sulfate, sodium sulfate, sodium sulfate, sodium sulfate, sodium sulfate, sodium stearate, sodium stearate, and gelatin.
- Sterile injectable solutions may be prepared by incorporating the active compounds in the required amounts, in the appropriate solvent, with various of the other ingredients enumerated above, as required, followed by filtered sterilization.
- dispersions may be prepared by incorporating the sterilized active ingredient into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above.
- the preferred methods of preparation may include vacuum drying and the freeze drying technique that yields a powder of the active ingredient, plus any additional desired ingredient from the previously sterile- filtered solution thereof.
- the therapeutic compounds of this invention may be administered to a patient alone or in combination with a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier may be determined, for example, by the solubility and chemical nature of the compounds, chosen route of administration and standard pharmaceutical practice.
- the dosage of the compounds of the present invention that will be most suitable for prophylaxis or treatment will vary with the form of administration, the particular compound chosen and the physiological characteristics of the particular patient under treatment. Generally, small dosages may be used initially and, if necessary, increased by small increments until the desired effect under the circumstances is reached. Generally speaking, oral administration may require higher dosages.
- a daily dosage of the compound of the invention may range from about 0.001 to about 100 milligrams of the compound of the invention, preferably a compound as described herein, (and all combinations and subcombinations of ranges and specific dosage amounts therein), per kilogram of patient body weight.
- the daily dosage may be about 0.01 to about 10 milligrams of the compound of the invention, preferably a compound as described herein per kilogram of patient body weight.
- the daily dosage may be about 0.1 milligrams of the compound of the invention, preferably a compound as described herein per kilogram of patient body weight.
- the compounds of the invention preferably a compound as described herein, generally may be present in an amount of about 0.1 to about 4 milligrams.
- the combination products of this invention such as pharmaceutical compositions comprising cannabinoids and/or opioids in combination with the compounds of Formula I, Ia, II, III, or rv, may be in any dosage form, such as those described herein, and can also be administered in various ways, as described herein.
- the combination products of the invention are formulated together, in a single dosage form (that is, combined together in one capsule, tablet, powder, or liquid, etc.).
- the cannabinoid and/or opioid compounds and the compounds of Formula I, Ia, II, III, or IV may be administered at the same time (that is, together), or in any order.
- the administration of a cannabinoid and/or opioid and the compounds of Formula I, Ia, II, III, or IV occurs less than about one hour apart, more preferably less than about 30 minutes apart, even more preferably less than about 15 minutes apart, and still more preferably less than about 5 minutes apart.
- administration of the combination products of the invention is oral, although other routes of administration, as described above, are contemplated to be within the scope of the present invention.
- the cannabinoids and/or opioids and the compounds of Formula I, Ia, II, III, or IV are both administered in the same fashion (that is, for example, both orally), if desired, they may each be administered in different fashions (that is, for example, one component of the combination product may be administered orally, and another component may be administered intravenously).
- the dosage of the combination products of the invention may vary depending upon various factors such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration, the age, health and weight of the recipient, the nature and extent of the symptoms, the kind of concurrent treatment, the frequency of treatment, and the effect desired.
- a daily dosage may range from about 0.01 to about 100 milligrams of the cannabinoid and/or opioid (and all combinations and subcombinations of ranges therein) and about 0.001 to about 100 milligrams of the compounds of Formula I, Ia, II, III, or IV (and all combinations and subcombinations of ranges therein), per kilogram of patient body weight.
- the a daily dosage may be about 0.1 to about 10 milligrams of the cannabinoid and/or opioid and about 0.01 to about 10 milligrams of the compounds of Formula I, Ia, II, III, or IV per kilogram of patient body weight. Even more preferably, the daily dosage may be about 1.0 milligrams of the cannabinoid and/or opioid and about 0.1 milligrams of the compounds of Formula I, Ia, II, III, or IV per kilogram of patient body weight.
- a typical dosage form of this type of combination product such as a tablet
- the cannabinoid compounds e.g.
- ⁇ 9 -tetrahydrocannabinol or cannabidiol and/or the opioid compounds (e.g., morphine) and generally may be present in an amount of about 15 to about 200 milligrams, and the compounds of Formula I, Ia, II, III, or IV in an amount of about 0.1 to about 4 milligrams.
- the preferred dosage forms of the combination products of this invention are formulated such that although the active ingredients are combined in a single dosage form, the physical contact between the active ingredients is minimized (that is, reduced).
- one embodiment of this invention where the product is orally administered provides for a combination product wherein one active ingredient is enteric coated.
- enteric coating one or more of the active ingredients it is possible not only to minimize the contact between the combined active ingredients, but also, it is possible to control the release of one of these components in the gastrointestinal tract such that one of these components is not released in the stomach but rather is released in the intestines.
- Another embodiment of this invention where oral administration is desired provides for a combination product wherein one of the active ingredients is coated with a sustained-release material that effects a sustained-release throughout the gastrointestinal tract and also serves to minimize physical contact between the combined active ingredients.
- the sustained-released component can be additionally enteric coated such that the release of this component occurs only in the intestine.
- Still another approach would involve the formulation of a combination product in which the one component is coated with a sustained and/or enteric release polymer, and the other component is also coated with a polymer such as a low- viscosity grade of hydroxypropyl methylcellulose (HPMC) or other appropriate materials as known in the art, in order to further separate the active components.
- HPMC hydroxypropyl methylcellulose
- the polymer coating serves to form an additional barrier to interaction with the other component.
- Dosage forms of the combination products of the present invention wherein one active ingredient is enteric coated can be in the form of tablets such that the enteric coated component and the other active ingredient are blended together and then compressed into a tablet or such that the enteric coated component is compressed into one tablet layer and the other active ingredient is compressed into an additional layer.
- one or more placebo layers may be present such that the placebo layer is between the layers of active ingredients.
- dosage forms of the present invention can be in the form of capsules wherein one active ingredient is compressed into a tablet or in the form of a plurality of microtablets, particles, granules or non-perils, which are then enteric coated. These enteric coated microtablets, particles, granules or non-perils are then placed into a capsule or compressed into a capsule along with a granulation of the other active ingredient.
- the amount of the compound, or an active salt or derivative thereof, required for use in treatment will vary not only with the particular salt selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will be ultimately at the discretion of the attendant physician or clinician.
- the desired dose may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day.
- the sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations; such as multiple inhalations from an insufflator or by application of a plurality of drops into the eye.
- the dose may also be provided by controlled release of the compound, by techniques well known to those in the art.
- kits useful in, for example, the treatment of pain which comprise a therapeutically effective amount of a cannabinoid and/or opioid along with a therapeutically effective amount of a pyridine compound of the invention, in one or more sterile containers, are also within the ambit of the present invention. Sterilization of the container may be carried out using conventional sterilization methodology well known to those skilled in the art.
- the sterile containers of materials may comprise separate containers, or one or more multi-part containers, as exemplified by the UNIVIALTM two-part container (available from Abbott Labs, Chicago, Illinois), as desired.
- the opioid or cannabinoid compound and the compound of Formula I, II, III, or IV may be separate, or combined into a single dosage form as described above.
- kits may further include, if desired, one or more of various conventional pharmaceutical kit components, such as for example, one or more pharmaceutically acceptable carriers, additional vials for mixing the components, etc., as will be readily apparent to those skilled in the art.
- kit components such as for example, one or more pharmaceutically acceptable carriers, additional vials for mixing the components, etc., as will be readily apparent to those skilled in the art.
- Instructions, either as inserts or as labels, indicating quantities of the components to be administered, guidelines for administration, and/or guidelines for mixing the components, may also be included in the kit.
- the compounds, pharmaceutical compositions and methods of the present invention may involve a peripheral cannabinoid receptor agonist compound.
- peripheral designates that the compound acts primarily on physiological systems and components external to the central nervous system.
- the peripheral receptor agonist compounds employed in the methods of the present invention exhibit high levels of activity with respect to peripheral tissue, such as, gastrointestinal tissue, while exhibiting reduced, and preferably substantially no CNS activity.
- substantially no CNS activity means that less than about 20% of the pharmacological activity of the compounds employed in the present methods is exhibited in the CNS, preferably less than about 15%, more preferably less than about 10%, even more preferably less than about 5%, and most preferably 0%, of the pharmacological activity of the compounds employed in the present methods is exhibited in the CNS.
- the compounds of the present invention may be used in methods to bind cannabinoid receptors, more preferably CB1 or CB2 cannabinoid receptors. Such binding may be accomplished by contacting the receptor with an effective amount of a compound of Formula I, II, III, or IV.
- the cannabinoid receptors may be located in the central nervous system or located peripherally to the central nervous system or in both locations.
- the contacting step conducted in an aqueous medium, preferably at physiologically relevant ionic strength, pH, and the like.
- the compound of the invention administered does not substantially cross the blood-brain barrier and thereby reduces the classical central side effects as observed for blood-brain penetrating cannabinoid agonists such as ⁇ 9 -tetrahydrocannabinol ( ⁇ 9 -THC).
- blood-brain penetrating cannabinoid agonists such as ⁇ 9 -tetrahydrocannabinol ( ⁇ 9 -THC).
- ⁇ 9 -THC blood-brain penetrating cannabinoid agonists
- the central side effects of blood brain penetrating cannabinoid agonists limits their clinical utility, such as their use in the relief of pain.
- does not substantially cross means that less than about 30% by weight of the compound employed in the present methods crosses the blood-brain barrier, preferably less than about 15% by weight, more preferably less than about 10% by weight, even more preferable less than about 5% by weight and most preferably 0% by weight of the compound crosses the blood-brain barrier.
- Selected compounds can be evaluated for CNS penetration by determining plasma and brain levels following i.v. administration.
- the invention is directed to methods of binding cannabinoid receptors, preferably CB1 and/or CB2 receptors, comprising the step of administering to a patient in need thereof, an effective amount of a compound of the invention including, for example, a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
- the methods comprise the step of administering to said patient an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
- the cannabinoid receptors are CB1 and/or CB2 cannabinoid receptors.
- the compound selectively binds CB2 cannabinoid receptors relative to CB1 receptors, even more preferably peripheral CB2 receptors.
- the cannabinoid receptors are located in the central nervous system. In other preferred embodiments, the cannabinoid receptors are located peripherally to the central nervous system. In some other preferred embodiments, the compound exhibits activity toward the cannabinoid receptors. In certain preferred embodiments, the binding agonizes the activity of the cannabinoid receptors.
- the binding antagonizes the activity of the cannabinoid receptors. In still other preferred embodiments, the binding inversely agonizes the activity of the cannabinoid receptors. [0152] In certain embodiments, the present invention is directed to methods of treating a gastrointestinal disorder, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or FV, or any combination thereof.
- the gastrointestinal disorders which may be treated with the present compounds and methods include, for example, nausea, vomiting, loss of appetite, cachexia, diarrhoea, inflammatory bowel disease, or irritable bowel syndrome, or any combination thereof.
- the present invention is directed to methods of treating inflammation, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
- the present invention is directed to methods of treating autoimmune diseases, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or FV, or any combination thereof.
- the autoimmune disease is multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, systemic lupus erythematosus, myasthenia gravis, diabetes mellitus type I, osteoporosis, or any combination thereof.
- the present invention is directed to methods of treating an immune related disorder, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
- the immune related disorder is asthma, chronic pulmonary obstructive disorder, emphysema, bronchitis, allergy, tissue rejection in organ transplants, celiac disease, or Sjogren's syndrome, or any combination thereof.
- the present invention is directed to methods of treating pain, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or FV, or any combination thereof.
- the pain may be inflammatory pain, neuropathic pain, visceral pain, surgical pain, including pain which occurs during surgery or pain which occurs after surgery (i.e., postsurgical pain), or cancer related pain.
- the present pain ameliorating methods may further comprise the administration to the patient of at least one opioid in the form of combination products and/or combination therapy.
- Suitable opioids include, for example, alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil or tramadol, and mixtures thereof.
- the present methods may further comprise administering to a patient codeine, carbamazepine, gabapentin, lamotrigine, phenytoin, amitryptiline, an NMDA receptor antagonist, an ion channel antagonist, or a nicotinic receptor agonist, or a mixture thereof, in the form of combination products and/or combination therapy.
- the methods for treating pain may further comprise the administration to the patient of at least one cannabinoid.
- the present invention is directed to methods of treating hypertension, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
- the present invention is directed to methods of treating neurodegenerative diseases, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
- the neurodegenerative disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, or amyotrophic lateral sclerosis.
- these methods may further comprise the administration to the patient of deprenyl, amantadine, levodopa, or carbidopa, in the form of combination products and/or combination therapy.
- the present invention is directed to methods of treating neurological disorders, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or FV, or any combination thereof.
- the neurological disorder is stroke, migraine, or cluster headache, or any combination thereof.
- the present invention is directed to methods of providing cardioprotection against ischemic and reperfusion effects, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
- the ischemic or reperfusion effect is arrhythmia or hypertension, or a combination thereof.
- the present invention is directed to methods of inhibiting mechanical hyperalgesia associated with nerve injury, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
- the present invention is directed to methods of inducing apoptosis in malignant cells, comprising the step of contacting said cells with an effective amount of a compound of Formula I, Ia, II, III, or FV, or any combination thereof.
- the apoptosis occurs in vitro, In other preferred embodiments, the apoptosis occurs in vivo.
- the present invention is directed to methods for modulating appetite, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
- the modulating decreases appetite.
- the modulating enhances appetite.
- pyridine compounds of Formula I, Ia, II, III, or IV can be readily prepared.
- the invention is further described in the following examples.
- the actual examples, herein provided, are for illustrative purposes only, and are not to be construed as limiting the appended claims. They provide a series of pyridine derivatives (IA-I ID) of Formula I, Ia, II, III, or IV, prepared according to Schemes 1-25, shown below in Examples 1 A-25G.
- the prophetic examples 12, 13, and any intermediates thereto, herein provided in Schemes 12 and 13, are for illustrative purposes only, and are not to be construed as limiting the appended claims.
- Schemes 12 and 13 are prophetic.
- Commercially available 5,6,7,8- tetrahydroisoquinoline 12.1 is oxidized with hydrogen peroxide (Gribble et al., Tetrahedron 1988, 44(11), 3195-3202) and chlorinated with phosphorous oxychloride (Yamanaka et al., Chemical & Pharmaceutical Bulletin 1988, 36(6), 2244-7) to yield 12.2.
- Conversion to 12.3 is accomplished following a procedure described by Mac Bride et al., Synthetic Commun. 1996, 2(5(12), 2309-2316.
- a Wittig reaction followed by hydroboration applicable to the synthesis of 12.5 is described in WO20040052851.
- Compounds 14AE - 14AG were prepared using methane thiolate c10, ethane thiolate ell and acid chlorides a3, al4 or a2 respectively.
- Compound 15A was prepared as outlined in Scheme 15. Commercially available methyl 2-amino-6-methylisonicotinate 15.1 was brominated, converted to amide 15.3 and reduced to 15.4 with borane dimethyl sulfide complex. The 4-bromo substituent in 15.4 was replaced through a reaction with sodium methoxide and copper bromide to give 15.5, followed by amide formation with acid chloride a2 to yield compound 15 A.
- Compound 21 A - 21D were prepared as outlined in Scheme 21.
- Intermediate 2.3a2 was reacted with triethyl phosphate.
- the phosphonate 21.1 was reacted under Horner Wadsworth Emmons conditions with 1-acetylpiperidin-4-one or dihydro-2H-pyran-4(3H)-one.
- Intermediate 21.2a or 21.2c was reacted with methyl zinc chloride and reduced to compounds 21 A or 21C.
- Intermediate 20.4 was reacted with triethyl phosphate, coupled with 1- acetylpiperidin-4-one and reduced to compound 21B.
- the aqueous layer was basified with aqueous NaOH, and extracted with DCM. The organic layers were combined, dried over Na 2 SO 4 and concentrated in vacuo. The residue was purified by column chromatography using acetone-hexane (1:2) as eluent.
- N,7V-diiso-propylethylamine (260 ⁇ L, 0.0015 mol) was added at once to the ice cooled reaction and after 1 hour morpholine (320 ⁇ L, 0.0037 mol) was added at once and the reaction was allowed to slowly warm to room temperature and stir overnight.
- the reaction was diluted with 35 mL of ethyl acetate and washed with 25 mL of water. The aqueous phase was back extracted with 25 mL of ethyl acetate and the combined extracts were dried with sodium sulfate and concentrated under vacuum.
- the material was chromatographed on a 12 gram silica gel column with a gradient of 15 to 50 % ethyl acetate/hexanes.
- the oil obtained was dissolved in 4 mL of methanol and 1.5 mL of HCl in dioxane was added. After ten minutes the solvents were removed under vacuum and the sample dried overnight. The yield was 123 mg (32%).
- N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide 2A was prepared from 2.3 and morpholine bl following general method C in 36% yield.
- Example 2C was prepared from 2 A following general method E in 20% yield.
- Examples 2D - 2E were prepared from 2A using 4-pyridine boronic acid c3 and ethyl boronic acid c4 respectively following general method E.
- Examples 2F - 21 were prepared from the appropriate starting materials and following general methods as described in examples 2A and 2B.
- the vial was swept with nitrogen for ten minutes and sodium-tert-butoxide (0.070 g, 0.73 mmol) and 2-methoxy-ethylamine (c6, 1.5 mL, 0.017 mol) were added.
- the reaction was heated in the microwave for 60 minutes at 100 °C. LC/MS indicated the desired product was formed as the major product and starting material was still present.
- the reaction was heated at 115 °C for 70 minutes.
- the reaction was partitioned in 30 mL of water and 50 mL of ethyl acetate.
- the aqueous phase was extracted with an additonal 50 mL of ethyl acetate.
- the combined organic extracts were dried with sodium sulfate and concentrated under vacuum.
- Examples 2L - 2R were prepared from the appropriate starting materials and following general methods as described in examples 2A and 2B.
- Examples 2T - 2V were prepared from the appropriate starting materials and following general methods as described in example 2A.
- Example 3B was prepared from 3.1 and acid chloride a4 following general method A.
- Examples 3C - 3H were prepared from 3.1 and the appropriate acid chloride following general method A.
- Examples 31 - 3O were prepared from 3.1 and acids el- e9 utilizing the coupling reagent Bop-Cl (bis(2-oxo-3-oxazolidinyl)phosphinic chloride).
- N-(5-bromo-4-(bromomethyl)-6-methylpyridin-2-yl)-2,2-dimethyl-butanamide 5.3 and N-(5-bromo-6-(bromomethyl)-4-methylpyridin-2-yl)-2,2-dimethylbutanamide 5.4 were obtained as a mixture when N-(5-bromo-4,6-dimethylpyridin-2-yl)-2,2-dimethyl-butanamide 5.2 was reacted with NBS/AIBN following general procedure B. A separation at this stage was not possible.
- Example 5B was also obtained from this reaction (2.0 g, 61% yield).
- Example 5D 2,2-dimethyl-N-(6-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide, was obtained as a minor product (28% yield) from the same reaction.
- N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide 5E was obtained from 5B according to general method D in 85% yield.
- N-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide 6 A was prepared from 6.2 according to general method B, followed by general method C in 65% overall yield.
- Example 6B was obtained from 6A in 76% yield following general method D.
- Example 8B was obtained from 8.4 and 2-methylcyclohexanecarbonyl chloride al following general method A in 76% yield.
- Example 10C was prepared in a similar fashion to 10A using 2-butanone d3 and acid chloride al .
- Examples 10D - 10L were prepared in a similar fashion to 10A using 10.7dl or 10.7dx and the appropriate acid chloride.
- Examples 10N - 10O were prepared in a similar fashion to 10M using 10.6dl and 1- methoxypropan-2-amine or (1R,2R,4S)-bicyclo[2.2.1]heptan-2-amine respectively.
- Examples 11C and 11D were prepared from 2B and 10C following the procedure described for example 11B.
- Example 14B was prepared from 2A and c7 following the procedure described for example 14 A.
- Example 14 was prepared from 14B and a4 following e procedures described for example 3A.
- Example 14D - 14K were prepared from 2A and the appropriate acid chlorides following the procedures described for example 3A.
- Examples 14M - 14S were prepared from 14.2a and the appropriate acids following the procedure described for example 14L.
- Compound 14T was prepared from 14.2b and acid chloride a2 following general method A. MS analysis m/z - 335+1.
- Examples 14U - 14AD were prepared from 14.2b and the appropriate acid chlorides or acids following the procedure described for example 14T or 14L respectively.
- Example 14AF was prepared from 14AE following the procedures described for example 14C.
- Example 14AG was prepared from 2A and sodium ethane thiolate and acid chloride a2 following the procedures described for example 14T.
- a 5 mL microwave vial was charged with the t-butyl carbamate (141 mg, 1.21 mmol), xantphos (30 mg, 0.06 mmol) , cesium carbonate (441 mg, 1.35 mmol) and tris(dibenzylideneacetone)dipalladium (0) Pd 2 (dba) 3 (20 mg, 0.02 mmol) and the vial was flushed with nitrogen for ten minutes.
- the 2-iodo-4-(morpholinomethyl)-5-propoxypyridine (16.6, 350 mg, 0.97 mmol) was dissolved in 1,4-dioxane (1.5 mL) and added as a solution.
- the reaction was flushed with nitrogen for an additional ten minutes and then heated at 70 °C overnight.
- the reaction was diluted with 30 mL of water and extracted with two 60 mL portions of ethyl acetate.
- the organic phase was dried with sodium sulfate and concentrated under vacuum.
Abstract
Novel pyridine compounds, pharmaceutical compositions containing the pyridine compounds, and methods of their pharmaceutical use are disclosed. In certain embodiments, the pyridine compounds are agonists and/or ligands of cannabinoid receptors and may be useful, inter alia, for treating and/or preventing pain, gastrointestinal disorders, inflammation, auto-immune diseases, ischemic conditions, immune-related disorders, hypertension, neurological disorders, and neurodegenerative diseases, for providing cardioprotection against ischemic and reperfusion effects, for inducing apoptosis in malignant cells, for inhibiting mechanical hyperalgesia associated with nerve injury, and as an appetite stimulant.
Description
PYRIDINE COMPOUNDS AND METHODS OF THEIR USE
FIELD OF THE INVENTION
[0001] This invention relates to novel pyridine compounds, pharmaceutical compositions containing such compounds, and uses thereof. More particularly, the present invention relates to novel pyridine compounds that may affect the cannabinoid receptor system and thus may be useful, inter alia, as agonists or antagonists of cannabinoid receptors.
BACKGROUND OF THE INVENTION
[0002] Cannabis sativa preparations have long been known as therapeutic agents to treat various diseases (Mechoulam, R., "Cannabinoids as Therapeutic Agents" CRC Press, Boca Raton, FIa. 1-19, 1986). The native active constituent, delta 9-tetrahydrocannabinol (Δ9-THC), is prescribed today, under the generic name dronabinol, as an anti-emetic and for enhancement of appetite, mainly in AIDS patients. However, separation between the clinically undesirable psychotropic effects and the therapeutically desirable effects on the peripheral nervous systems, the cardiovascular system, the immune and endocrine system is problematic. The discovery of two cannabinoid receptors, CB1 and CB2, has helped to elucidate the diverse cannabinoid effects.
[0003] The CB1 receptor has been cloned from rat, mouse, and human tissues and exhibits 97- 99% amino acid sequence identity across species. The CB2 receptor exhibits 48% homology with the CB1 receptor (A.C. Howlett et al. Pharmacological Reviews 2002, 54, 161-202). The structures of both receptors are consistent with seven transmembrane G-protein coupled receptors. In addition, both receptors exert their effect by negative regulation of adenylyl cyclase activity through pertussis toxin-sensitive GTP-binding proteins. They have been shown to activate the mitogen activated protein kinase (MAPK) in certain cell types (Parolaro, D., Life Sci. 1999, 65, 637-44).
[0004] The CB1 receptor is expressed mainly in the central nervous systems (CNS) and to a lesser extent in other tissues including, for example, gastrointestinal tissues, immune cells, reproductive organs, heart, lung, urinary bladder, and adrenal gland. The CB2 receptor is expressed mostly in peripheral tissue associated with immune functions including, for example, macrophages, B cells, T cells and mast cells, as well as in peripheral nerve terminals (Perrwee,
R.G., Prog. Neurobiol. 2001, 63, 569-611). The central distribution pattern of CB1 receptors accounts for several unwanted pharmacological properties of cannabinoids, such as impaired cognition and memory, altered control of motor function, and psychotropic and other neurobehavioral effects. CB1 receptors are also found on pain pathways in brain, spinal cord and at the peripheral terminals of primary sensory neurons (A. S. Rice, Curr. Opin. Investig. Drugs 2001 2(3), 399-414).
[0005] CB1 knockout mice have been shown to be unresponsive to cannabinoids in behavioral assays providing molecular evidence that the psychotropic effects, including sedation, hallucinations, and antinociception are manifested through the activation of the CB1 receptor, present primarily in the CNS. Analysis of the CB2 knockout mouse has corroborated the evidence for the function of CB2 receptors in modulating the immune system. The CB2 receptor does not affect immune cell development and differentiation as determined by FACS analysis of cells from the spleen, lymph node and thymus from CB2 knockout mice. Further studies in these mice have shown that the immunosuppressive effects of Δ9-THC are mediated by the CB2 receptor.
[0006] Some cannabinoid receptor agonists have been shown to produce potent antinociception with equivalent efficacy to morphine in animal models of acute pain, persistent inflammatory pain, and neuropathic pain. They have also been reported to induce a number of unwanted CNS side effects. Furthermore, the known cannabinoid receptor agonists are in general highly lipophilic and insoluble in water. There is thus a need for cannabinoid receptor agonists with improved properties for uses as therapeutic agents.
[0007] Known CB1 cannabinoid receptor agonists produce a characteristic profile of in vivo effects in mice, including suppression of spontaneous activity, antinociception, hypothermia, and catalepsy. Measurement of these four properties, commonly referred to as the tetrad test, has played a key role in establishing the structure-activity relationship of cannabinoids and cannabimimetics acting at CB1 receptors. Catalepsy in mice is indicative of CB1 activation and predictive of cannabinoid psychoactivity. Pertwee showed a correlation between catalepsy in the ring test in mice and the previously validated dog static ataxia model (R.G. Pertwee, Br. J. Pharmacology 1972, 46, 753-763). Therefore, catalepsy in mice is viewed as an excellent predictor of CNS effects in humans (D.R. Compton, Marijuana: An International Research
Report 7, 213-218, 1987; E.W. Gill and G. Jones, Biochem. Pharmacol. 21, 2237-2248, 1972; E.W. Gill et al. Nature 228, 134-136, 1970).
[0008] Efforts have been made to separate therapeutic effects from undesirable CNS side effects by increasing the selectivity for the CB2 receptor, thereby leading to efforts to design compounds with selectivity for the CB2 receptor over the CB1 receptor. These compounds would be predicted to lack side effects even if they penetrate the CNS because they would not activate the CB1 receptors in the CNS (Malan, T. Philip, Jr. et al. "CB2 cannabinoid receptor agonists: pain relief without psychoactive effects?" Curr Op. Pharm. 2003, 3(1), 62-67; WO2004/017920).
[0009] Recent studies have identified CB2 selective inverse agonists with antiedema effects in vivo (Iwamura et al., J. Pharm. Exp. Ther. 2001, 420-425; Lavey et al. Bioorg. Med. Chem. Lett. 2005, 783-786), suggesting an involvement of CB2 selective inverse agonists in inflammatory processes and the pharmacological efficacy of CB2 inverse agonists by themselves.
[0010] Lunn, et al. (ICRS 15th Annual Symposium of the Cannabinoids, Clearwater Beach, FL, June 24-27, 2005) have reported that CB2 receptor-selective inverse agonists are capable of altering cellular chemotaxis mediated by either cannabinoids or chemokines, both in vivo and in vitro. They have reported that administration of these compounds can decrease allergic eosinophilia in animal models for asthma.
[0011] Other research has shown that pharmacological antagonists of CB1 and CB2 receptors prevented ovariectomy-induced bone loss in vivo and caused osteoclast inhibition in vitro by promoting osteoclast apoptosis and inhibiting production of several osteoclast survival factors. These studies show that the CB1 receptor has a role in the regulation of bone mass and ovariectomy-induced bone loss and that CB1- and CB2-selective cannabinoid receptor antagonists are a new class of osteoclast inhibitors that may be of value in the treatment of osteoporosis and other bone diseases (A. I. Idris, et al., Nature Medicine, 2005,77(7), 774-79).
[0012] There is considerable interest in developing new cannabimimetic compounds possessing preferentially high affinity for the CB2 receptor. Such compounds that preferentially stimulate the CB2 receptor, directly or indirectly, may provide clinically useful effects without unwanted effects on the subject's central nervous system and can offer a rational therapeutic
approach to a variety of disease states. The present invention is directed to these and other important ends.
SUMMARY OF THE INVENTION
[0013] Accordingly, the present invention is directed, in part, to novel pyridine compounds which may be modulators, agonists, and/or antagonists of cannabinoid receptors and which thus may be useful, inter alia, for the treatment of diseases or disorders which are associated with the cannabinoid receptor system.
[0014] Specifically, in certain embodiments, the present invention relates to compounds of formula I:

A is a ring atom or a bond;
D is cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, Or -NR2R3; E is N or CR12; Y is N or CR6;
R1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -ORx, -SRx, -NRyRz, F, Cl, or Br, or R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R8)(R9)]n-D; provided that: when R1 or D independently includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7-membered ring having 0 or 1 heteroatom ring members, and
when R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic ring, then the formed monocyclic ring has 0 or 1 heteroatom ring members;
Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl; or Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
R2 and R3 are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl, or R2 and R3 when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
Z is -C(=O)-, -N(R5)-,-C(=O)N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-;
R4 is:

R5 and R5a are each independently H or alkyl;
Rc is H, alkyl, or aryl;
R and Re are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
R6 and R12 are each independently H, F, Cl, or alkyl, or R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R8)(R9)]n-D;
R7 is H, F, or alkyl, or R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R7 and R12 taken together with the atoms through
which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
R and R are each independently H or alkyl; each R9a is independently H, alkyl, aryl, -C(=O)-R11, -C(=O)-OR11, -[C(R11)(R11)]s-C(=O)-OR11, -SO2R11, or -C(=O)N(R11)RU; each R10 is independently H, alkyl, or aryl; each R11 is independently H or alkyl; n and r are each independently 0, 1, 2, or 3; and s is 1, 2, 3, or 4; provided that:
(1) only one of E and Y is N;
(2) at least one of R1, R6, and R7 is other than F;
(3) when R7 is F, then at least one of R1 and R6 is other than F, Cl, or Br;
(4) when R1 is -ORX, then E is N;
(5) when n is 0, then -A-[C(R8)R9)]n-D is other than aryl or heteroaryl;
(6) at least one of R2 and R3 is other than H;
(7) at least two of Rc, Rd, and Re are other than H; and
(8) no more than one pair of R7 and R12, R1 and R7 , R1 and A, and R6 and A form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
[0015] In certain embodiments, the invention is directed to compounds according to formula Ia:

wherein:
A is -S(=O)2- or a bond;
D is -N(H)-S(=O)2-aryl, -N(Rv)-S(=O)2-aryl, -N(H)aralkyl, -N(CH3)aralkyl, or heterocycloalkyl, in which the heterocycloalkyl ring contains at least one nitrogen atom or dioxo-thio group; provided that: when A is -S(=O)2-, then n is 0 and D is -N(H)aralkyl, -N(CH3)aralkyl, or heterocycloalkyl, wherein the heterocycloalkyl group in D contains at least one nitrogen atom, and A is attached to D through the heterocycloalkyl nitrogen ring atom; and when A is a bond, then D is -N(H)-S(=O)2-aryl, -N(Rv)-S(=O)2-aryl, or a heterocycloalkyl ring containing at least one dioxo-thio group;
E is N or CR12;
Y is N or CR6;
R1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -ORX, -SRx, -NRyRz, F, Cl, or Br; provided that when R1 includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members,
Rv is C1-3unsubstituted alkyl;
Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl; or Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
Z is -N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-;
R4 is:

R6 and R12 are each independently H, F, Cl, or alkyl;
R7 is H, F, or alkyl, or R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
R8 and R9 are each independently H or alkyl; each R9a is independently H, alkyl, aryl, -C(=O)-R11, -C(=O)-ORn, -[C(R11)(R11)]s-C(=O)-OR1 1, -SO2R11, or -C(=O)N(R11)R11; each R10 is independently H, alkyl, or aryl; each R11 is independently H or alkyl; n and r are each independently 0, 1, 2, or 3; and s is 1, 2, 3, or 4; provided that:
(1) only one of E and Y is N;
(2) at least two of Rc, Rd, and Re are other than H; and
(3) no more than one pair of R7 and R12, and R1 and R7 form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
[0016] The present invention is directed, in part, to pharmaceutical compositions, comprising: a pharmaceutically acceptable carrier and a compound of formula I or Ia.
[0017] The present invention is also directed, in part, to methods of binding cannabinoid receptors in a patient in need thereof, comprising the step of administering to said patient an effective amount of a compound according to formula I or Ia.
DETAILED DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
[0018] The present invention is generally directed to pyridine compounds, pharmaceutical compositions containing these compounds, and methods of their pharmaceutical use.
[0019] As employed above and throughout the disclosure, the following terms, unless otherwise indicated, shall be understood to have the following meanings.
[0020] As used herein, the term "alkyl" refers to an optionally substituted, saturated, straight or branched hydrocarbon having from about 1 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), preferably with from about 1 to about 8 carbon atoms, more preferably with from about 1 to about 6 carbon atoms, yet more preferably with from about 1 to about 4 carbon atoms, still more preferably with from about 1 to about 3 carbon atoms, with 1 carbon being even more preferred. In some preferred embodiments, 1 or more of the hydrogen atoms on the alkyl group, more preferably from 1 to about 6, still more preferably about 3 to about 6, yet more preferably all of the hydrogen atoms on one or two of the terminal methyl moieties of the alkyl group are substituted with fluorine atoms. Alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl, 3-methylpentyl, 2,2-dimethylbutyl, and 2,3-dimethylbutyl.
[0021] As used herein, the term "cycloalkyl" or "carbocyclic ring " each refers to an optionally substituted, mono-, di-, tri-, or other multicyclic alicyclic ring system having from about 3 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein). In some preferred embodiments, the cycloalkyl groups have from about 3 to about 8 carbon atoms, more preferably from about 3 to about 6 carbon atoms. Multi-ring structures may be bridged or fused ring structures, wherein the additional groups fused or bridged to the cycloalkyl ring may include optionally substituted cycloalkyl, aryl, heterocycloalkyl, or heteroaryl rings. Exemplary cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl, adamantyl, 2-[4-isopropyl-1- methyl-7-oxa-bicyclo[2.2.1]heptanyl], and 2-[1,2,3,4-tetrahydro-naphthalenyl].
[0022] As used herein, "bicycloalkyl" refers to an optionally substituted, alicyclic group having two bridged rings in its structure and having from about 7 to about 20 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 7 to about 15 carbon atoms being preferred. Exemplary bicycloalkyl-ring structures include, but are not limited to, norbornyl, bornyl, [2.2.2]-bicyclooctyl, cis-pinanyl, trans-pinanyl, camphanyl, iso-bornyl, and fenchyl.
[0023] As used herein, "tricycloalkyl" refers to an optionally substituted, alicyclic group having three bridged rings in its structure and having from about 7 to about 20 carbon atoms (and
all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 7 to about 15 carbon atoms being preferred. Exemplary tricycloalkyl-ring structures include, but are not limited to, tricyclo[5.1.2.02,6]decane, 1,7,7-trimethyl tricyclo[2.2.1.02,6]heptane, alpha-santalol, patchouli alcohol, alpha-cedrene, and longifolene.
[0024] As used herein, the term "cycloalkylalkyl" refers to an optionally substituted ring system comprising an alkyl radical bearing one or more cycloalkyl substituents, and having from about 6 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 6 to about 10 carbon atoms being preferred, wherein alkyl and cycloalkyl are as previously defined. Non-limiting examples include, for example, cyclopropylmethyl, cyclobutylethyl, cyclopentylpropyl, cyclohexylmethyl, 2-cyclooctyl-1-methylethyl, 2-[4-isopropyl-1-methyl-7-oxa-bicyclo[2.2.1]heptanyl]methyl, 2-[1,2,3,4-tetrahydro-naphthalenyl]ethyl, and adamantylpropyl.
[0025] As used herein, the term "alkylcycloalkyl" refers to an optionally substituted ring system comprising a cycloalkyl group having one or more alkyl substituents, wherein cycloalkyl and alkyl are each as previously defined. Exemplary alkylcycloalkyl groups include 2-methylcyclohexyl, 3,3-dimethylcyclopentyl, trans-2,3-dimethylcyclooctyl, and 4-methyldecahydronaphthalenyl.
[0026] As used herein, the term "heterocycloalkyl" and "heterocyclic ring" each refers to an optionally substituted ring system composed of a cycloalkyl radical wherein in at least one of the rings, one or more of the carbon atom ring members is independently replaced by a heteroatom group selected from the group consisting of O, S, N, and NH, wherein cycloalkyl is as previously defined. Heterocycloalkyl ring systems having a total of from about 5 to about 14 carbon atom ring members and heteroatom ring members (and all combinations and subcombinations of ranges and specific numbers of carbon and heteroatom ring members) are preferred. In other preferred embodiments, the heterocyclic groups may be fused to one or more aromatic rings. In certain preferred embodiments, heterocycloalkyl moieties are attached via a ring carbon atom to the rest of the molecule. Exemplary heterocycloalkyl groups include, but are not limited to, azepanyl, tetrahydrofuranyl, hexahydropyrimidinyl, tetrahydrothienyl, piperidinyl, pyrrolidinyl, isoxazolidinyl, isothiazolidinyl, pyrazolidinyl, oxazolidinyl, thiazolidinyl, piperazinyl, 2-oxo-morpholinyl, morpholinyl, 2-oxo-piperidinyl, piperadinyl, decahydroquinolyl, octahydrochromenyl, octahydro-cyclopenta[c]pyranyl, 1,2,3 ,4,-tetrahydroquinolyl,
1,2,3,4-tetrahydroquinazolinyl, octahydro-[2]pyridinyl, decahydro-cycloocta[c]furanyl, 1,2,3,4-tetrahydroisoquinolyl, 2-oxo-imidazolidinyl, and imidazolidinyl. In some embodiments, two moieties attached to a heteroatom may be taken together to form a heterocycloalkyl ring, such as when R and R , taken together with the nitrogen atom to which they are attached, form a heterocycloalkyl ring. In certain of these embodiments, 1 or 2 of the heterocycloalkyl ring carbon atoms may be replaced by other moieties which contain either one (-O-, -S-, -N(R9)-) or two (-N(R10)-C(=O)-, or -C(=O)-N(R10)-) ring replacement atoms. When a moiety containing one ring replacement atom replaces a ring carbon atom, the resultant ring, after replacement of a ring atom by the moiety, will contain the same number of ring atoms as the ring before ring atom replacement. When a moiety containing two ring replacement atoms replaces a ring carbon atom, the resultant ring after replacement will contain one more ring atom than the ring prior to replacement by the moiety. For example, when a piperidine ring has one of its ring carbon atoms replaced by -N(R10)-C(=O)-, the resultant ring is a 7-membered ring containing 2 ring nitrogen atoms and the carbon of a carbonyl group in addition to 4 other carbon ring atoms (CH2 groups) from the original piperidine ring.
[0027] As used herein, the term "heterocycloalkylalkyl" refers to an optionally substituted ring system composed of an alkyl radical having one or more heterocycloalkyl substituents, wherein heterocycloalkyl and alkyl are as previously defined. In some preferred embodiments, the alkyl moieties of the heterocycloalkylalkyl groups have from about 1 to about 3 carbon atoms. Exemplary heterocycloalkyl groups include, but are not limited to, azepanylmethyl, tetrahydrofuranylethyl, hexahydropyrimidinylisobutyl, tetrahydrothienylpropyl, piperidinyl-2,2- dimethylethyl, pyrrolidinylmethyl , isoxazolidinylethyl, isothiazolidinylpropyl, pyrazolidinylmethyl, oxazolidinylbutyl, thiazolidinylisopropyl, piperazinylmethyl, 2-oxo-morpholinylmethyl, morpholinylethyl, 2-oxo-piperidinylethyl, piperadinylmethyl, decahydroquinolylethyl, octahydrochromenylpropyl, octahydro-cyclopenta[c]pyranylbutyl, 1,2,3,4,-tetrahydroquinolylethyl, 1,2,3,4-tetrahydroquinazolinylmethyl, octahydro- [2]pyridinylethyl, decahydro-cycloocta[c]furanylmethyl, 1,2,3,4-tetrahydroisoquinolylmethyl, 2-oxo-imidazolidinylethyl, and imidazolidinylmethyl.
[0028] As used herein, the term "alkenyl" refers to an optionally substituted alkyl group having from about 2 to about 10 carbon atoms and one or more double bonds (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), wherein alkyl is as previously defined.
[0029] As used herein, the term "alkynyl" refers to an optionally substituted alkyl group having from about 2 to about 10 carbon atoms and one or more triple bonds (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), wherein alkyl is as previously defined.
[0030] As used herein, the term "aryl" refers to an optionally substituted, mono-, di-, tri-, or other multicyclic aromatic ring system having from about 6 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), preferably with from about 6 to about 10 carbons, with about 6 carbon atoms being preferred. Non-limiting examples include, for example, phenyl, naphthyl, anthracenyl, and phenanthrenyl. In certain preferred embodiments, aryl is phenyl, optionally substituted, more preferably substituted with at least one halo or haloalkyl group.
[0031] As used herein, the term "aralkyl" refers to an optionally substituted ring system comprising an alkyl radical bearing an aryl substituent and having from about 7 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), preferably with from about 7 to about 11 carbon atoms, with about 7 carbon atoms being preferred. Non-limiting examples include, for example, benzyl, diphenylmethyl, triphenylmethyl, phenylethyl, and diphenylethyl.
[0032] As used herein, the term "alkylaralkyl" refers to an optionally substituted ring system comprising an alkyl radical bearing an aralkyl substituent and having from about 8 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 8 to about 12 carbon atoms being preferred, wherein alkyl and aralkyl are as previously defined. Non-limiting examples include, for example, to lylm ethyl, bis(isopropylphenyl)methyl, 1-tolyl-1-ethylphenylmethyl, tert-butylphenylethyl, and ortΛo-methyl-jpαra-butylphenylethyl.
[0033] As used herein, the term "alkoxyl" refers to an optionally substituted alkyl-O- group wherein alkyl is as previously defined. In some preferred embodiments, the alkyl moieties of the alkoxy groups have from about 1 to about 4 carbon atoms. Exemplary alkoxy groups include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, and heptoxy.
[0034] As used herein, the term "aryloxyl" refers to an optionally substituted aryl-O- group wherein aryl is as previously defined. Exemplary aryloxy groups include, but are not limited to, phenoxy and naphthoxy.
[0035] As used herein, the term "aralkoxyl" refers to an optionally substituted aralkyl-O- group wherein aralkyl is as previously defined. Exemplary aralkoxy groups include, but are not limited to, benzyloxy, 1-phenylethoxy, 2-phenylethoxy, and 3-naphthytheptoxy.
[0036] As used herein, the term "1,1-dioxo-thio" refers to a diradical having the structure:

[0037] Preferably, a 1 , 1 -dioxo-thio is a ring member of a heterocyclic ring. Exemplary ring member 1,1-dioxo-groups such as 1,1-dioxo-thiirane, 1,1-dioxo-thietane, 1,1-dioxo- tetrahydrothiophene, 1,1-dioxo-thiopyran, 1,1-dioxo-thiepane, and the like, more preferably 1,1- dioxo-thiopyran. In certain embodiments one or more ring carbon atoms of a 1,1-dioxo-thio ring is optionally replaced by -O-, -S-, or -N(R9a)-. Exemplary rings include 1,1-dioxo- thiomorpholine and [1,4]oxathiane 4,4-dioxide.
[0038] As used herein, the term "halo" refers to a fluoro, chloro, bromo, or iodo moiety, with fluoro, chloro, or bromo moieties being preferred. In certain embodiments, fluoro or chloro moieties are more preferred.
[0039] As used herein, the term "heteroaryl" refers to an optionally substituted aryl ring system wherein in at least one of the rings, one or more of the carbon atom ring members is independently replaced by a heteroatom group selected from the group consisting of S, O, N, and NH, wherein aryl is as previously defined. Heteroaryl groups having a total of from about 5 to about 14 carbon atom ring members and heteroatom ring members(and all combinations and subcombinations of ranges and specific numbers of carbon and heteroatom ring members) are preferred. Exemplary heteroaryl groups include, but are not limited to, pyrryl, furyl, pyridyl, pyridine-N-oxide, 1,2,4-thiadiazolyl, pyrimidyl, thienyl, isothiazolyl, imidazolyl, tetrazolyl, pyrazinyl, pyrimidyl, quinolyl, isoquinolyl, thiophenyl, benzothienyl, isobenzofuryl, pyrazolyl, indolyl, purinyl, carbazolyl, benzimidazolyl, and isoxazolyl. Heteroaryl may be attached via a carbon or a heteroatom to the rest of the molecule.
[0040] As used herein, the term "heteroarylalkyl" refers to an optionally substituted ring system comprising an alkyl radical bearing a heteroaryl substituent, having from about 2 to about 50 carbon atoms (and all combinations and subcombinations of ranges and specific numbers of carbon atoms therein), with from about 6 to about 25 carbon atoms being preferred. Non- limiting examples include 2-(1H-pyrrol-3-yl)ethyl, 3-pyridylmethyl, 5-(2H-tetrazolyl)methyl, and 3 -(pyrimidin-2-yl)-2-methylcyclopentanyl.
[0041] As used herein, the term "spiroalkyl" refers to an optionally substituted alkylene diradical, both ends of which are bonded to the same carbon atom of the parent group to form a spirocyclic group. The spirocyclic group, as herein defined, has 3 to 20 ring atoms, preferably with 3 to 10 ring atoms. Exemplary spiroalkyl groups taken together with its parent group include, but are not limited to, 1-(1-methyl-cyclopropyl)-propan-2-one, 2-(1-phenoxy- cyclopropyl)-ethylamine, and 1-methyl-spiro[4.7]dodecane.
[0042] Typically, substituted chemical moieties include one or more substituents that replace hydrogen. Exemplary substituents include, for example, halo (e.g., F, Cl, Br, I), alkyl, cycloalkyl, alkylcycloalkyl, alkenyl, alkynyl, aralkyl, aryl, heteroaryl, heteroarylalkyl, spiroalkyl, heterocycloalkyl, hydroxyl (-OΗ), alkoxyl, aryloxyl, aralkoxyl, nitro (-NO2), cyano (-CN), amino (-NH2), N-substituted amino (-NHR"), N,N-disubstituted amino (-N(R")R"), carboxyl (-COOH), -C(=O)R", -OR", -C(=O)OR", -C(=O)NHSO2R", -NHC(=O)R", aminocarbonyl (-C(=O)NH2), N-substituted aminocarbonyl (-C(=O)ΝHR"), N,N-disubstituted aminocarbonyl (-C(=O)Ν(R")R"), thiolato (SR"), sulfonic acid and its esters (-SO3R"), phosphonic acid and its mono-ester (-P(=O)(OR")(OH) and di-esters (-P(=O)(OR")(OR"), -S(=O)2R", -S(=O)2NH2, -S(=O)2NHR", -S(=O)2NR"R", -SO2NHC(=O)R", -NHS(=O)2R", -NR"S(=O)2R", -CF3, -CF2CF3, -NHC(=O)NHR", -NHC(=O)NR"R", -NR"C(=O)NHR", -NR"C(=O)NR"R", -NR"C(=O)R" and the like. In relation to the aforementioned substituents, each moiety R" can be, independently, any of H, alkyl, cycloalkyl, alkenyl, aryl, aralkyl, heteroaryl, or heterocycloalkyl, or when (R"(R")) is attached to a nitrogen atom, R" and R" can be taken together with the nitrogen atom to which they are attached to form a 4- to 8-membered nitrogen heterocycle, wherein the heterocycloalkyl ring is optionally interrupted by one or more additional -O-, -S-, -SO, -SO2-, -NH-, -N(alkyl)-, or -N(aryl)- groups, for example, In certain embodiments, chemical moieties are substituted by at least one optional substituent, such as those provided hereinabove. In the present invention, when chemical moieties are substituted with optional susbstituents, the optional substituents are not further substituted. For example,
when R1 is an alkyl moiety, it is optionally substituted, based on the definiton of "alkyl" as set forth herein. Specifically, when R1 is alkyl substituted with optional aryl, the optional aryl substituent is not further substituted. To further clarify, 2-(alpha-naphthyl)ethyl (wherein ethyl is the alkyl moiety and alpha-naphthyl is the optional aryl substituent) is within the scope of optionally substituted alkyl. In contrast, 2-(3-chlorophenyl)ethyl (wherein ethyl is the alkyl moiety and 3-chlorophenyl is the optional substituent) is not within the scope of optionally substituted alkyl because the optional aryl substiuent cannot be further substituted by a further chemical group.
[0043] As used herein, the term "cannabinoid" refers to any one of a group of naturally occurring compounds of related structure that may be isolable from Cannabis sativa, more commonly known as marijuana, and structurally modified derivatives thereof. Cannabinoids include for example, compounds such as Δ9-tetrahydrocannabinol, Δ8-tetrahydrocannabinol, cannabichromene, cannabicyclol, cannabidiol, cannabielsoin, cannabigerol, cannabinol, cannabitriol, nabilone, and nantradol, and numerous structural variants. Typically cannabinoids are lipophilic and have low solubility in water.
[0044] As used herein, the term "cannabimimetic" refers to any of a group of endogenous or exogenous receptor ligands that bind one or more of the receptors bound by cannabinoids and mimic one or more behaviors of cannabinoids while so bound. Examples of endogenous cannabimimetics (also referred to as "endocannabinoids") produced in mammalian tissues include, for example, arachidonoylethanolamide (anandamide), 2-arachidonoyl glycerol, 1(3)- arachidonoyl glycerol, and palmitoylethanolamide. Examples of exogenous cannabimimetics include, for example WIN 55,212-2, CP 55,940, HU-210, and the like. Other examples of exogenous cannbimimetics may be found in publications such as R.B. Pertwee, "Pharmacology of Cannabinoid Receptor Ligands", Current Medicinal Chemistry, 1999, 6, 635-664, and A.C. Howlett, et al. "International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors", Pharmacological Reviews, 2002, 54(2), 161-202, the disclosures of which are each hereby incorporated herein by reference in their entireties.
[0045] As used herein, the term "antagonist" refers to a compound that binds to a receptor to form a complex that preferably does not elicit any response, in the same manner as an unoccupied receptor, and does not alter the equilibrium between inactive and active receptor.
[0046] As used herein, "agonist" refers to a ligand that produces a conformational change in the receptor and alters the equilibrium of the receptor's active and inactive states, which in turn induces a series of events, resulting in a measurable biological response. Agonists include, for example, conventional agonists, which exhibit positive receptor activity, and inverse agonists, which exhibit a negative intrinsic activity.
[0047] As used herein, the term "prodrug" refers to compounds that may serve to maximize the amount of active species that reaches the desired site of reaction that are themselves typically inactive or minimally active for the activity desired, but through biotransformation are converted into biologically active metabolites.
[0048] As used herein, the term "stereoisomers" refers to compounds that have identical chemical constitution, but differ as regards the arrangement of the atoms or groups in space.
[0049] As used herein, the term "partial stereoisomers" refers to stereoisomers having two or more chiral centers wherein at least one of the chiral centers has defined stereochemistry (i.e., R or S) and at least one has undefined stereochemistry (i.e., R or S). When the term "partial stereoisomers thereof is used herein, it refers to any compound within the described genus whose configuration at chiral centers with defined stereochemistry centers is maintained and the configuration of each undefined chiral center is independently selected from R or S. For example, if a stereoisomer has three chiral centers and the stereochemical configuration of the first center is defined as having "S" stereochemistry, the term "or partial stereoisomer thereof refers to stereoisomers having SRR, SRS, SSR, or SSS configurations at the three chiral centers, and mixtures thereof.
[0050] Asymmetric carbon atoms may be introduced into the molecule depending on the structure of the moiety R4 when Ra and Rb are non-identical or when Rc, Rd, and Re are non- identical. For example, when Ra is hydrogen and Rb is other than H, the carbon atom to which Ra is attached is asymmetric.
[0051] Other asymmetric centers are contemplated in the present invention. Asymmetric centers are, by convention, present in R4 moieties structure such as those shown below at the ring carbon atoms identified with an asterisk (*). As such, these classes of compounds can exist as

the individual "R" or "S" stereoisomers at each or any of these asymmetric centers, alone or in combination with any other asymmetric centers so formed in the compound to provide single enantiomers, any of the possible racemic mixtures of isomers or diastereomeric mixtures thereof, and all are contemplated as within the scope of the present invention.
[0052] As used herein, the term "N-oxide" refers to compounds wherein the basic nitrogen atom of either a heteroaromatic ring or tertiary amine is oxidized to give a quaternary nitrogen bearing a positive formal charge and an attached oxygen atom bearing a negative formal charge.
[0053] As used herein, the term "hydrate" refers to a compound of the present invention which is associated with water in the molecular form, i.e., in which the H-OH bond is not split, and may be represented, for example, by the formula RΗ2O, where R is a compound of the invention. A given compound may form more than one hydrate including, for example, monohydrates (R.H2O) or polyhydrates (R.nH2O wherein n is an integer > 1) including, for example,
dihydrates (R.2H2O), trihydrates (R' 3H2O), and the like, or hemihydrates, such as, for example,
R.n/2H2O, R.n/3H2O, R.n/4H2O and the like wherein n is an integer.
[0054] As used herein, the term "solvate" refers to a compound of the present invention which is associated with solvent in the molecular form, i.e., in which the solvent is coordinatively bound, and may be represented, for example, by the formula Resolvent), where R is a compound of the invention. A given compound may form more than one solvate including, for example, monosolvates (R. (solvent)) or polysolvates (R.n(solvent)) wherein n is an integer > 1) including,
for example, disolvates (R.2(solvent)), trisolvates (R.3(solvent)), and the like, or hemisolvates,
such as, for example, R.n/2(solvent), R. n/3 (solvent), R.n/4(solvent) and the like wherein n is an
integer. Solvents herein include mixed solvents, for example, methanol/water, and as such, the solvates may incorporate one or more solvents within the solvate.
[0055] As used herein, the term "acid hydrate" refers to a complex that may be formed through association of a compound having one or more base moieties with at least one compound having one or more acid moieties or through association of a compound having one or more acid moieties with at least one compound having one or more base moieties, said complex being further associated with water molecules so as to form a hydrate, wherein said hydrate is as previously defined and R represents the complex herein described above.
[0056] As used herein, the term "pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like. These physiologically acceptable salts are prepared by methods known in the art, e.g., by dissolving the free amine bases with an excess of the acid in aqueous alcohol, or neutralizing a free carboxylic acid with an alkali metal base such as a hydroxide, or with an amine.
[0057] Compounds described herein throughout, can be used or prepared in alternate forms. For example, many amino-containing compounds can be used or prepared as an acid addition salt. Often such salts improve isolation and handling properties of the compound. For example, depending on the reagents, reaction conditions and the like, compounds as described herein can be used or prepared, for example, as their hydrochloride or tosylate salts. Isomorphic crystalline forms, all chiral and racemic forms, N-oxide, hydrates, solvates, and acid salt hydrates, are also contemplated to be within the scope of the present invention.
[0058] Certain acidic or basic compounds of the present invention may exist as zwitterions. All forms of the compounds, including free acid, free base and zwitterions, are contemplated to be within the scope of the present invention. It is well known in the art that compounds containing both basic nitrogen atom and acidic groups often exist in equilibrium with their zwitterionic forms. Thus, any of the compounds described herein throughout that contain, for example, both basic nitrogen and acidic groups, also include reference to their corresponding zwitterions.
[0059] As used herein, the term "effective amount" refers to an amount of a compound as described herein that may be therapeutically effective to inhibit, prevent or treat the symptoms of particular disease, disorder or side effect. Such diseases, disorders and side effects include, but are not limited to, those pathological conditions associated with the binding of cannabinoid receptors (for example, in connection with the treatment and/or prevention of pain), wherein the treatment or prevention comprises, for example, agonizing the activity thereof by contacting cells, tissues or receptors with compounds of the present invention. Thus, for example, the term "effective amount," when used in connection with cannabinoids, for example, for the treatment of pain, refers to the treatment and/or prevention of the painful condition. The term "effective amount", when used in connection with the present cannabinoid receptor agonist compounds, refers to the treatment, reduction and/or prevention of side effects typically associated with cannabinoids including, for example, such side effects as those hereinabove mentioned.
[0060] As used herein, the term "pharmaceutically acceptable" refers to those compounds, materials, compositions, and/or dosage forms that are, within the scope of sound medical judgment, suitable for contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problems or complications commensurate with a reasonable benefit/risk ratio.
[0061] As used herein, the term "in combination with", "combination therapy" and "combination products" refer, in certain embodiments, to the concurrent administration to a patient of cannabinoids and the compounds of the Formula I, II, III, or IV. When administered in combination, each component may be administered at the same time or sequentially in any order at different points in time. Thus, each component may be administered separately but sufficiently closely in time so as to provide the desired therapeutic effect.
[0062] As used herein, the term "dosage unit" refers to physically discrete units suited as unitary dosages for the particular individual to be treated. Each unit may contain a predetermined quantity of active compound(s) calculated to produce the desired therapeutic effect(s) in association with the required pharmaceutical carrier. The specification for the dosage unit forms of the invention may be dictated by (a) the unique characteristics of the active compound(s) and the particular therapeutic effect(s) to be achieved, and (b) the limitations inherent in the art of compounding such active compound(s).
[0063] The term "treatment" as used herein includes preventative (e.g., prophylactic), curative or palliative treatment and "treating" as used herein also includes preventative, curative and palliative treatment.
[0064] As used herein, the term "pain" refers to the perception or condition of unpleasant sensory or emotional experience, which may or may not be associated with actual or potential tissue damage or described in terms of such damage. "Pain" includes, but is not limited to, two broad categories of pain: acute and chronic pain (Buschmann, H.; Christoph, T; Friderichs, E.; Maul, C; Sundermann, B; eds.; Analgesics, Wiley- VCH, Verlag GMbH & Co. KgaA, Weinheim; 2002; Jain, K. K. "A Guide to Drug Evaluation for Chronic Pain"; Emerging Drugs, 5(2), 241-257(2000)). Non-limiting examples of pain include nociceptive pain, inflammatory pain, visceral pain, somatic pain, neuropathic pain, AIDS pain, cancer pain, phantom pain, and psychogenic pain, and pain resulting from hyperalgesia, pain caused by rheumatoid arthritis, migraine, allodynia, and the like.
[0065] As used herein, the term "patient" refers to animals, including mammals, preferably humans.
[0066] As used herein, the term "side effect" refers to a consequence other than the one(s) for which an agent or measure is used, as the adverse effects produced by a drug, especially on a tissue or organ system other then the one sought to be benefited by its administration. In the case, for example, of cannabinoids, the term "side effect" may refer to such conditions as, for example, psychotropic effects, such as confusion, anxiety, panic, distortion of perception, fantasizing, sedation, inner unrest, irritability and insomnia, sweating, rhinorrhoea, loose stools, hiccups, dry mouth, tachycardia, ataxia, dizziness, orthostatic hypotension, and anorexia.
[0067] When any variable occurs more than one time in any constituent or in any formula, its definition in each occurrence is independent of its definition at every other occurrence. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
[0068] It is believed the chemical formulas and names used herein correctly and accurately reflect the underlying chemical compounds. However, the nature and value of the present invention does not depend upon the theoretical correctness of these formulae, in whole or in part. Thus it is understood that the formulas used herein, as well as the chemical names attributed to the correspondingly indicated compounds, are not intended to limit the invention in any way, including restricting it to any specific tautomeric form or to any specific optical or geometric isomer, except where such stereochemistry is clearly defined.
[0069] Accordingly, the present invention is directed, in part, to a new class of cannabinoid receptor modulator compounds, preferably pyridine compounds, which may be highly useful in connection with the binding of cannabinoid receptors. Compounds binding cannabinoid receptors may agonize and/or antagonize the receptors. In situations where a cannabimimetic compound or ligand agonizes one or more cannabinoid receptors, the resultant binding is believed to trigger an event or series of events in the cell that results in a change in the cell's activity, its gene regulation, or the signals that it sends to neighboring cells, similar to that of a cannabinoid. Thus, in some embodiments, compounds of the invention may serve to prevent or treat diseases or disorders in which cannabinoid receptors are implicated. In situations where a cannabimimetic compound or ligand antagonizes one or more cannabinoid receptors, the resultant binding typically occurs comparatively to a greater extent relative to that of the cannabinoid, but does not trigger one or more of the events of signal transduction. Compounds with these properties are highly useful, for example, in connection with the study of functions of cannabinoid receptors, which may result, for example, in the development of new cannabimimetic agonist compounds, such as those, for example, reported in Rinaldi-Carmona, M. et al., Journal of Pharmacology and Experimental Therapeutics, 1998, 284(2), 644-650, the disclosure of which is hereby incorporated herein by reference, in its entirety.
[0070] Accordingly, in one embodiment, the present invention provides compounds of formula I:
 wherein:
wherein:
A is a ring atom or a bond;
D is cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, Or -NR2R3; E is N or CR12; Y is N or CR6;
R1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -ORx, -SRX, -NRyRz, F, Cl, or Br, or R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R8)(R9)]n-D; provided that: when R1 or D independently includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7-membered ring having 0 or 1 heteroatom ring members, and when R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic ring, then the formed monocyclic ring has 0 or 1 heteroatom ring members; Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl; or Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, Or-C(=O)-N(R10)-;
R2 and R3 are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl, or R2 and R3 when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
Z is -C(=O)-, -N(R5)-,-C(=O)N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-;
R4 is:
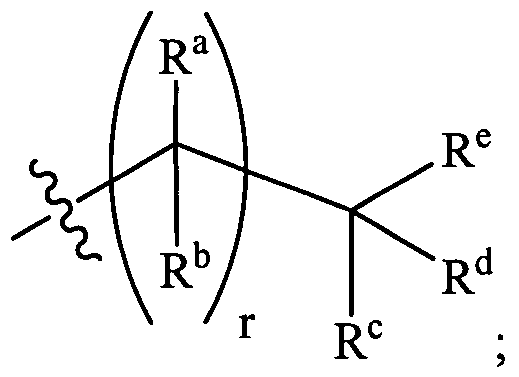
R5 and R5a are each independently H or alkyl;
Rc is H, alkyl, or aryl;
Rd and Re are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
R6 and R12 are each independently H, F, Cl, or alkyl, or R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R8)(R9)]n-D;
R7 is H, F, or alkyl, or R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
R8 and R9 are each independently H or alkyl; each R9a is independently H, alkyl, aryl, -C(=O)-Rn, -C(=O)-OR11, -[C(R11)(R11)]s-C(=O)-OR11, -SO2R11, or -C(=O)N(R11)R11; each R10 is independently H, alkyl, or aryl; each R11 is independently H or alkyl; n and r are each independently 0, 1, 2, or 3; and s is 1, 2, 3, or 4; provided that:
(1) only one of E and Y is N ( i.e., at least one of, but no more than one of E and Y is N);
(2) at least one of R1, R6, and R7 is other than F;
(3) when R7 is F, then at least one of R1 and R6 is other than F, Cl, or Br;
(4) when R1 is -ORX, then E is N;
(5) when n is 0, then -A-[C(R8)R9)]n-D is other than aryl or heteroaryl;
(6) at least one of R2 and R3 is other than H;
(7) at least two of Rc, Rd, and Re are other than H; and
(8) no more than one pair of R7 and R12, R1 and R7 , R1 and A, and R6 and A form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
In other embodiments, the present invention provides compounds of formula Ia:

A is -S(=O)2- or a bond;
D is -N(H)-S(=O)2-aryl, -N(Rv)-S(=O)2-aryl, -N(H)aralkyl, -N(CH3)aralkyl, or heterocycloalkyl, in which the heterocycloalkyl ring contains at least one nitrogen atom or dioxo-thio group; provided that: when A is -S(=O)2-, then n is 0 and D is -N(H)aralkyl, -N(CH3)aralkyl, or heterocycloalkyl, wherein the heterocycloalkyl group in D contains at least one nitrogen atom, and A is attached to D through the heterocycloalkyl nitrogen ring atom; and when A is a bond, then D is -N(H)-S(=O)2-aryl, -N(Rv)-S(=O)2-aryl, or a heterocycloalkyl ring containing at least one dioxo-thio group;
E is N or CR 112.
Y is N or CR6;
R1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -ORX, -SRX, -NRyRz, F, Cl, or Br; provided that when R1 includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members,
Rv is C1-3unsubstituted alkyl;
Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl; or Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
Z is -N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-;
R4 is:

R5 and R5a are each independently H or alkyl;
Rc is H, alkyl, or aryl;
R and Re are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
R6 and R12 are each independently H, F, Cl, or alkyl;
R7 is H, F, or alkyl, or R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
R8 and R9 are each independently H or alkyl; each R9a is independently H, alkyl, aryl, -C(=O)-R11, -C(=O)-OR11, -[C(R11)(R1 1)]s-C(=O)-OR11, -SO2R11, or -C(=O)N(R1 ^R11J each R10 is independently H, alkyl, or aryl;
each R i l l is independently H or alkyl; n and r are each independently 0, 1, 2, or 3; and s is 1, 2, 3, or 4; provided that:
(1) only one of E and Y is N;
(2) at least two of Rc, Rd, and Re are other than H; and
(3) no more than one pair of R7 and R12, and R1 and R7 form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
[0072] In certain other preferred embodiments, the compound of formula I or Ia has the structure of formula II:

wherein R1, R4, R5, R6, R7, R8, R9, A, D, and n are as defined above.
[0073] Alternatively preferred, the compound of formula I or Ia has the structure of formula III:
 wherein R1, R4, R7, R8, R9, A, D, and n are as defined above.
wherein R1, R4, R7, R8, R9, A, D, and n are as defined above.
[0074] In other certain preferred embodiments, the compound of formula I has the structure of formula IV:

[0075] In certain preferred embodiments of the compounds of formula I, Ia, II, or III, A is a bond.
[0076] In other preferred embodiments of compounds of the formula Ia, A is -S(=O)2-.
[0077] In certain preferred embodiments of the compounds of formula I, Ia, II, III, or IV, D is -NR2R3.
[0078] In other preferred embodiments of the compounds of formula Ia, D is -N(H)-S(=O)2- aryl or -N(Rv)-S(=O)2-aryl.
[0079] Alternatively, in certain preferred embodiments of compounds of the formula Ia, D is heterocycloalkyl containing at least one nitrogen atom or dioxo-thio group; more preferably heterocycloalkyl containing at least one nitrogen atom. In embodiments wherein D is heterocycloalkyl containing at least one nitrogen atom, D is preferably morpholinyl, piperidinyl, or 1,1-dioxo-thiomorpholinyl, each optionally substituted.
[0080] In still other preferred embodiments of compounds of the formula Ia, D is N(H)-aralkyl or -N(CH3)-aralkyl.
[0081] In some preferred embodiments of the compounds of formula I, Ia, or IV, E is N.
[0082] In other preferred embodiments of the compounds of formula I, Ia, or IV, Y is N.
[0083] In certain preferred embodiments of the compounds of formula I, Ia, II, or III, R1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, hetero aralkyl, -ORX, -SRX, -NRyRz, F, Cl, or Br, more preferably H, alkyl, cycloalkyl, aryl, heteroaryl, -ORX, or Br, with alkyl, -ORX, or Br being even more preferred. In some preferred embodiments, R1 is H. In other preferred embodiments, R1 is -ORX, more preferably alkoxy. In still other preferred embodiments, R1 is alkyl. In certain other preferred embodiments, when R1 includes a cycloalkyl, aryl, heterocycloalkyl or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members.
[0084] As used herein, the term "included ring moiety" refers to when any element independently, for example, R1, is a group having a ring moiety within the group. For example, in certain embodiments R1 is the group cycloalkyl, cycloalkylalkyl, aryl, aralkyl,
heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl. These groups respectively comprise the ring moiety cycloalkyl, cycloalkyl, aryl, aryl, heterocycloalkyl, heterocycloalkyl, heteroaryl, or heteroaryl. By way of further explanation, groups such as aralkyl, heterocycloalkylalkyl, and heteroaralkyl, which are alkyl moieties substituted with an aryl, heterocycloalkyl, and heteroaryl ring moieties respectively, have an included ring moiety.
[0085] In some preferred embodiments of the compounds of formula I, II, or III, R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 7-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R )(R )]n-D, and may be further optionally substituted. By way of further explanation, when R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group, the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic ring system, wherein the monocyclic ring group is substituted with -[C(R8)(R9)]n-D. In certain preferred embodiments, the -[C(R8)(R9)]n-D moiety is attached to the monocyclic ring carbon atom which is both adjacent to, and closer in proximity to the Y moiety in the core pyridine ring in the compound of formula I, II, or III. For example, when R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 6-membered aryl ring, the resulting fused bicyclic ring system which is formed will be a quinoline group or isoquinoline group, depending upon which of E and Y is N. When Y is N, the resulting fused bicyclic ring system is a substituted quinoline group. When E is N, the resulting fused bicyclic ring system is a substituted isoquinoline group. In certain preferred embodiments, when R1 and the ring atom A taken together with the atoms through which they are connected form a ring, then the formed monocyclic ring group has 0 or 1 heteroatom ring members.
[0086] In certain preferred embodiments of the compounds of formula I, Ia, II, or III, Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl. In other preferred embodiments, Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a 3- to 8-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -Ct=O)-N(R10)-. In still other preferred embodiments, Rx is alkyl.
[0087] In still other preferred embodiments of the compounds of formula I, Ia, II, III, or IV, R2 and R3 are each independently alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl, more preferably when at least one of R2 and R3 is aralkyl or cycloalkyl; still more preferably when at least one of R2 and R3 is aralkyl, with when one of R2 and R3 is aralkyl and the other is alkyl being even more preferred. In some embodiments, R3 is H or alkyl. Alternatively, R2 and R3 when taken together with the nitrogen atom to which they are attached, form a 3- to 7-membered, preferably 5- to 7-membered, heterocycloalkyl ring, wherein 1 or 2, preferably 1, of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(RIO)-C(=O)-, or -C(=O)-N(R10)-, more preferably wherein 1 or 2, preferably 1, of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, or -N(R9a)-, still more preferably when one of the heterocycloalkyl ring carbon atoms is replaced by -O-, -S-, or - N(R9a)-, with -O- being even more preferred. In certain embodiments wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a heterocycloalkyl ring, the ring is fused to a C6-aryl ring. In certain other embodiments wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a heterocycloalkyl ring, the heterocycloalkyl ring is substituted with at least one hydroxy, alkyl, dialkylamino, halo, heteroarylcarbonyl, or alkylcarbonyl group. In still other alternative embodiments, R2 and R3 are each independently alkyl.
[0088] In certain embodiments of the compounds of formula I, Ia, or IV, Z is -C(=O)-, -N(R5)-, -C(=O)N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-; preferably -C(=O)-, -C(=O)N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-; more preferably -C(=O)N(R5)- or -N(R5)C(=O)-; with -N(R5)C(=O)- being even more preferred. Alternatively, Z is - -N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-; more preferably-N(R5)-, or -N(R5)C(=O)-; yet more preferably -N(R5)-.
[0089] In some preferred embodiments of the compounds of formula I, Ia, II, III, or FV, R4 is :


[0091] In some preferred embodiments of the compounds of formula I, Ia, II, or IV, R5 is H. In other preferred embodiments of the compounds of formula I or IV, R5a is H.
[0092] In some embodiments of the compounds of formula I, Ia, II, III, or FV, each Ra and each Rb are independently H or alkyl. In certain preferred embodiments, each Ra is H. In other preferred embodiments, each Rb is H. In other more preferred embodiments, Ra and Rb are each H.
[0093] In still other preferred embodiments of the compounds of formula I, Ia, II, or FV, R6 is H.
[0094] In certain preferred embodiments of the compounds of formula I or II, R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 8-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R8)(R9)]n-D, and may be further optionally substituted . By way of further explanation, when R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group, the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic bicyclic ring system, wherein the monocyclic ring group is substituted with -[C(R8)(R9)]n-D. In certain preferred embodiments, the -[C(R8)(R9)]n-D moiety is attached to the monocyclic ring carbon atom which is either on a ring carbon alpha or beta to the core pyridine ring, and closer in proximity to the R1 moiety in the compound of formula I or
II. For example, when R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 6-membered aryl ring, the resulting fused bicyclic ring system which is formed will be an isoquinoline group. In certain preferred embodiments, when R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic ring, then the formed monocyclic ring group has 0 or 1 heteroatom ring members.
[0095] In certain embodiments of the compounds of formula I, Ia, II, III, or IV, Rc is H, alkyl, or aryl, preferably H or alkyl, with alkyl being more preferred. In some preferred embodiments where Rc is substituted alkyl, Rc is preferably alkoxy substituted alkyl. In other preferred embodiments where Rc is substituted aryl, Rc is preferably alkoxy substituted aryl.
[0096] In yet other preferred embodiments of the compounds of formula I, Ia, II, III, or IV, Rd and Re are each independently H or alkyl, more preferably alkyl. In certain more preferred embodiments, Rc, Rd, and Re are each independently alkyl. In some preferred embodiments where Rd or Re is substituted alkyl, it is more preferably alkoxy substituted alkyl.
[0097] In some preferred embodiments of the compounds of formula I, Ia, II, III, or IV, Rd and Re taken together with the carbon atom to which they are attached form a carbocyclic ring, preferably a monocyclic carbocyclic ring. In some embodiments, the carbocyclic ring is further substituted, preferably with at least one alkyl or alkoxy group, or any combination thereof.
[0098] In other preferred embodiments of the compounds of formula I, Ia, II, III, or IV, Rd and Re, taken together with the carbon atom to which they are attached, form a 3- to 12-membered carbocyclic ring, wherein the carbocyclic ring is substituted with 0-5 groups each independently selected from d.C4alkyl and C1.C4alkoxyl. More preferably, the carbocyclic ring is bicycloalkyl or tricycloalkyl, still more preferably bicycloalkyl; with bicycloalkyl ring is substituted with 1-3 alkyl, preferably
 groups.
groups.
[0099] In certain preferred embodiments of the compounds of formula I, Ia, II, III, or IV, R7 is H or alkyl. In other preferred embodiments, R7 is F.
[0100] In some preferred embodiments of the compounds of formula I, Ia, II, or III, R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7- membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 7-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available
position with -[C(R8)(R9)]n-D, and may be further optionally substituted . By way of further explanation, when R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group, the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic ring system. For example, when R1 and R7 taken together with the atoms through which they are connected form a monocyclic 6-membered aryl ring, the resulting fused bicyclic ring system which is formed will be a quinoline group or isoquinoline group, depending upon which of E and Y is N. When Y is N, the resulting fused bicyclic ring system is an isoquinoline group. When E is N, the resulting fused bicyclic ring system is a quinoline group. In certain preferred embodiments, when R1 and R7 taken together with the atoms through which they are connected form a ring, then the formed monocyclic ring group has 0 or 1 heteroatom ring members. In other more preferred embodiments, R1 and R7 taken together with the atoms through which they are attached form an aryl ring, even more preferably a C6aryl ring.
[0101] In still other preferred embodiments of the compounds of formula I, Ia, or IV, R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8- membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, more preferably a monocyclic 4- to 8-membered cycloalkyl ring, wherein the monocyclic ring group is substituted at any available position with -[C(R8)(R9)]n-D, and may be further optionally substituted . By way of further explanation, when R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring group, the result is a monocyclic ring group which is fused to the core pyridine ring to provide a fused bicyclic ring system. For example, when R7 and R12 taken together with the atoms through which they are connected form a monocyclic 6-membered aryl ring, the resulting fused bicyclic ring system which is formed will be an isoquinoline group. In certain preferred embodiments, when R7 and R12 taken together with the atoms through which they are connected form a ring, then the formed monocyclic ring group has 0 or 1 heteroatom ring members.
[0102] In certain embodiments of compounds of formula I. Ia, II, III, or IV, R and R are each H. Alternatively, they are each independently alkyl.
[0103] In some preferred embodiments of the compounds of formula I, Ia, or IV, R12 is H.
[0104] In some embodiments of the compounds of formula I, Ia, II, III, or IV, n and r are each independently 0, 1, 2, or 3, preferably 0, 1, or 2, more preferably 0 or 1. In certain even more preferred embodiments r is 0. In other even more preferred embodiments, n is 1.
[0105] In certain embodiments, the compound of formula I is selected from the group consisting of:
2-methyl-7V-(4-(moφholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide;
N-(5-bromo-4-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(moφholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-isopropyl-4-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(4-(moφholinomethyl)-5-phenyl-pyridin-2-yl)-butanamide;
N-(5-cyclohexyl-4-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-(furan-3-yl)-4-(moφholinomethyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(piperidin-1-ylmethyl)-pyridin-2-yl)-butanamide;
N-(4-((-N'-(2-methoxyethyl)-N'-(methyl)amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide ;
2-methyl-N-(5-methyl-4-(moφholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide; l,2-dimethyl-N-(5-methyl-4-(moφholinomethyl)-pyridin-2-yl)-cyclohexanecarboxamide;
2,2,3,3-tetramethyl-N-(5-methyl-4-(moφholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide; l,2,2,3,3-pentamethyl-N-(5-methyl-4-(moφholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
N-(5-bromo-6-methyl-4-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5,6-dimethyl-4-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(6-methyl-4-(moφholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-bromo-4-methyl-6-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(4,5-dimethyl-6-(moφholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
NN-(5-bromo-6-(m φholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-6-(moφholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-methyl-6-(moφholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-moφholinoquinolin-2-yl)-butanamide;
2-methyl-N-(4-moφholinoquinolin-2-yl)-cyclohexanecarboxamide;
2,2-dimethyl-N-(4-(moφholinomethyl)quinolin-2-yl)-butanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-6-(trifluoromethyl)pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(5-morpholino-5,6,7,8-tetrahydroisoquinolin-3-yl)-butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
[0106] More preferably, the compound is selected from the group consisting of:
2-methyl-N-(4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide;
N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-7V-(6-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(4-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-morpholinoquinolin-2-yl)-butanamide;
2-methyl-N-(4-morpholinoquinolin-2-yl)-cyclohexanecarboxamide;
2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
[0107] Still more preferably the compound of formula I is selected from the group consisting of:
N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(4-morpholinoquinolin-2-yl)-butanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
[0108] Even more preferred in some embodiments, the compound of formula I is selected from the group consisting of:
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
7V-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
7V-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
[0109] Alternatively, in certain embodiments the compound of formula I is selected from the group consisting of:
7V-(5-(2-methoxyethyl-amino)-4-(morpholinomethyl)-pyridin-2-yl)-pivalamide;
N-(5-bromo-4-(thiomorpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(thiomorpholinomethyl)-pyridin-2-yl)butanamide;
N-(4-((1,4-oxazepan-4-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-amino-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-4-((4-methylpiperazin-1-yl)methyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((4-acetylpiperazin-1-yl)methyl)-5-bromopyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-4-((4-phenylpiperazin-1-yl)methyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
1,2-dimethyl-7V-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)cyclohexanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide;
1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
1,2,2,3,3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclo- propanecarboxamide;
2,2-diethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide; 1-ethyl-7V-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclopropanecarboxamide;
3,3,3-trifluoro-2-methyl-7V-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)-2- (trifluoromethyl)propanamide;
2-cyclopropyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)propanamide;
2-cyclopropyl-2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)propanamide;
2-ethyl-2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclopentanecarboxamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)pentanamide;
4,4,4-trifluoro-2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide; 1-ethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclobutanecarboxamide; 1-ethyl-N-(4-(niθrpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclopentanecarboxamide; 1-ethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclohexanecarboxamide;
2,2-diethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)butanamide;
2,2,3,3-tetramethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]-pyridin-2- y^cyclopropanecarboxamide;
2-cyclopropyl-2-methyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]- pyridin-2-yl)propanamide;
N-(4-((1,4-oxazepan-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((1,4-oxazepan-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-1- ethylcyclopentanecarboxamide;
N-(4-((1,4-oxazepan-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-2,2- diethylbutanamide;
N-cycloheptyl-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-amine;
N-(1-methoxypropan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta-[b]pyridin- 2-amine;
N-(bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]- pyridin-2-amine;
N-(5-isopropyl-4-(morpholinomethyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2, 3,3- tetramethylcyclopropanecarboxamide;
N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-1,2,2, 3,3- pentamethylcyclopropanecarboxamide;
2-cyclopropyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2-methylpropanamide;
2-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2-methylbutanamide;
2,2-diethyl-7V-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)butanamide;
N-(5-ethyl-4-(morpholino-methyl)pyridin-2-yl)-3,3,3-trifluoro-2-methyl-2- (trifluoromethyl)propanamide; 1-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)cyclopentanecarboxamide; 1-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2,3,3- tetramethylcyclopropanecarboxamide;
2-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2-methylbutanamide;
2,2-diethyl-7V-(5-methoxy-4-(morpholinomethyl)-pyridin-2-yl)butanamide;
3,3,3-trifluoro-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2-methyl-2- (trifluoromethyl)-propanamide; 1-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)cyclopentanecarboxamide; 1-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2, 3,3- tetramethylcyclopropanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)- 1,2,2, 3,3- pentamethylcyclopropanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2-ethyl-2-methylbutanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-diethylbutanamide;
Ν-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-3,3,3-trifluoro-2-methyl-2- (trifluoromethyl)propanamide;
2-cyclopropyl-N-(5-ethoxy-4-(morpholinomethyl)-pyridin-2-yl)propanamide;
2-cyclopropyl-N-(5-ethoxy-4-(morpholinomethyl)-pyridin-2-yl)-2-methylpropanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-1-ethylcyclobutanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-1-ethylcyclopentanecarbox-amide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-1-ethylcyclohexanecarboxamide;
N-(5-(methylthio)-4-(morpholinomethyl)pyridin-2-yl)pivalamide; 1-ethyl-N-(5-(methylthio)-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide;
N-(5-(ethylthio)-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-methoxy-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
3,3,3-trifluoro-2-methyl-N-(4-(morpholinomethyl)-5-propoxypyridin-2-yl)-2- (trifluoromethyl)propanamide;
7V-(4-(isoindolin-2-y1-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((3,4-dihydroiso-quinolin-2(1H)-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((benzyl(ethyl)amino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((benzyl(methyl)-amino)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((benzyl(isopropyl)-amino)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-Λ/-(5-methyl-4-(pyrrolidin-1-ylmethyl)-pyridin-2-yl)butanamide;
N-(4-((3-hydroxypyrrolidin-1-yl)methyl)-5-methyl-pyridin-2-yl)-2,2-dimethyl- butanamide;
N-(4-((3-(dimethylamino)-pyrrolidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((3-fluoropyrrolidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(thiazolidin-3-ylmethyl)-pyridin-2-yl)butanamide;
N-(4-((2,5-dimethyl-2,5-dihydro-1H-pyrrol-1-yl)-methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-((4-methylpiperidinil-yl)methyl)pyridin-2-yl)-butanamide;
7V-(4-((3,5-dimethylpiperidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((4-hydroxypiperidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((3-fluoropiperidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-((4-methylpiperazin-1-yl)methyl)pyridin-2-yl)-butanamide;
N-(4-((4-ethylpiperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((4-acetylpiperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((bis(2-methoxyethyl)-amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-(((2-(dimethylamino)-ethyl)(ethyl)amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((sec-butylamino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((cyclopentylamino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((cyclohexylamino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-((2-methylcyclohexylamino)methyl)pyridin-2- yl)butanamide;
2,2-dimethyl-N-(5-methyl-4-((1-phenylethylamino)methyl)pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(5-methyl-4-(1-morpholinopropyl)-pyridin-2-yl)butanamide;
N-(4-((benzyl(methyl)-amino)methyl)-5-methylpyridin-2-yl)-2-cyclopropyl-2- methylpropanamide;
N-(2,3-dihydrobenzo-[b][1,4]dioxin-6-yl)-4-((2-(2,2-dimethylbutanamido)-5- methylpyridin-4-yl)-methyl)piperazine- 1 -carboxamide;
N-(4-((4-(1,2,3-thiadiazole-4-carbonyl)piperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2- cyclopropyl-2-methylpropanamide;
N-(4-(4-fluorophenethyl)-5,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5',6'-dimethyl-2,4'-bipyridin-2'-yl)-2,2-dimethylbutanamide;
N-(4-benzyl-5,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-4-phenylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-methoxy-4-phenylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((1-acetylpiperidin-4-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((1-acetylpiperidin-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-2,2- dimethylbutanamide ;
2,2-dimethyl-N-(5-methyl-4-((tetrahydro-2H-pyran-4-yl)methyl)pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(7-(4-(morpholinomethyl)-2,3-dihydrofuro-[3,2-b]pyridin-5- yl)butanamide; and
2,2-dimethyl-N-(5-methyl-4-(2-morpholinoethyl)-pyridin-2-yl)butanamide; or a pharmaceutically acceptable salt thereof.
[0110] Preferably, the compound of formula I is selected from the group consisting of:
N-((l/?,2i?,4S)-bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H- cyclopenta[b]-pyridin-2-amine;
(S)-N-(4-((3-fluoropyrrolidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide; and
(S)-2,2-dimethyl-N-(5-methyl-4-((1-phenylethylamino)methyl)pyridin-2-yl)-butanamide; or a pharmaceutically acceptable salt thereof.
[0111] Alternatively preferred, the compound of formula I is selected from the group consisting of:
1,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)cyclohexanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
1,2,2,3,3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2- yl)cyclopropanecarboxamide;
2,2-diethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide;
2-cyclopropyl-2-methyl-7V-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)propanamide; 1-ethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclohexanecarboxamide;
N-(bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]- pyridin-2-amine;
N-((li?,2i?,4-S)-bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H- cyclopenta[b]-pyridin-2-amine;
N-(5-ethyl-4-(morpholino-methyl)pyridin-2-yl)-2,2-dimethylbutanamide;
3,3,3-trifluoro-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2-methyl-2- (trifluoromethyl)-propanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
Ν-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-3,3,3-trifluoro-2-methyl-2- (trifluoromethyl)propanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-1-ethylcyclopentanecarboxamide; and
N-(4-(isoindolin-2-y1-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
[0112] In certain embodiments, the compound of formula Ia is selected from the group consisting of:
N-[4-(l , 1 -Dioxo- 1 -lambda *6*-thiomorpholin-4-yl-methyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyr amide;
N-[5-Bromo-4-(1,1-dioxo-1-lambda*6*-thiomorpholin-4-ylmethyl)-pyridin-2-yl]-3,3,3- trifluoro-2-methyl-2-trifluoromethylpropionamide;
N-[4-( 1 , 1 -Dioxo- 1 -lambda *6*-thiomorpholin-4-ylmethyl)-5 -methylpyridin-2-yl] -3 ,3 ,3 - trifluoro-2-methyl-2-trifluoromethylpropionamide;
2-Cyclopropyl-N-[4-(1,1-dioxo-1-lambda*6*-thiomorpholin-4-ylmethyl)-5- methylpyridin-2-yl]-isobutyramide;
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-yl-methyl)-5-methoxypyridin -2-yl]-2,2- dimethylbutyramide;
N-(5-bromo-4-((4-fluoro-N-methylphenylsulfonamido)methyl)pyridin-2-yl)-2,2- dimethylbutanamide;
N-(5-bromo-4-((N-methyl-4-(trifluoromethyl)phenylsulfonamido)methyl)pyridin-2-yl)- 2,2-dimethylbutanamide;
N-(4-((3-chloro-N-methylphenylsulfonamido)methyl)-5-methylpyridin-2-yl)-2- cyclopropyl-2-methylpropanamide;
2-cyclopropyl-N-(4-((4-fluoro-N-methylphenylsulfonamido)methyl)-5-methylpyridin-2- yl)-2-methylpropanamide;
2-cyclopropyl-2-methyl-N-(5-methyl-4-((N-methyl-4- (trifluoromethyl)phenylsulfonamido)methyl)pyridin-2-yl)propanamide;
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-ylmethyl)-5,6-dimethylpyridin-2-yl]- 2,2-dimethylbutyramide;
2-Cyclopropyl-N-[4-( 1,1 -dioxo- 1-lambda*6*-thiomorpholin-4-ylmethyl)-6,7-dihydro- 5H-[ 1 ]pyrindin-2-yl]-isobutyramide;
Λ/-[4-(1,1-Dioxo-hexahydro-1-thiopyran-4-ylmethyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyramide ;
2,2-dimethyl-N-(5-methyl-4-(morpholinosulfonyl)-pyridin-2-yl)butanamide; 2-cyclopropyl-2-methyl-Λ/-(5-methyl-4-(morpholinosulfonyl)pyridin-2-yl)-propanamide;
N-(5,6-dimethyl-4-(morpholinosulfonyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2-cyclopropyl-7V-(5,6-dimethyl-4-(morpholinosulfonyl)pyridin-2-yl)-2- methylpropanamide ;
2,2-dimethyl-N-(4-(morpholinosulfonyl)-6,7-dihydro-5H-cyclopenta-[b]pyridin-2- yl)butanamide;
2-cyclopropyl-2-methyl-N-(4-(morpholinosulfonyl)-6,7-dihydro-5H-cyclo- penta[b]pyridin-2-yl)propanamide; and
N-cyclohexyl-5,6-dimethyl-4-(moφholinosulfonyl)-pyridin-2-amine; or a pharmaceutically acceptable salt thereof.
[0113] Preferably, the compound of formula Ia is selected from the group consisting of:
N-[4-(l , 1-Dioxo- 1 -lambda *6*-thiomorpholin-4-yll-meth l)-5-methylpyridin-2-yl]-2,2- dimethylbutyramide; and
N-[4-(l , 1 -Dioxo- 1 -lambda *6*-thiomoφholin-4-ylmethyl)-5,6-dimethylpyridin-2-yl]-
2,2-dimethylbutyr amide; or a pharmaceutically acceptable salt thereof.
[0114] The compounds employed in the methods of the present invention may exist in prodrug form. As used herein, "prodrug" is intended to include any covalently bonded carriers which release the active parent drug, for example, as according to Formula I, Ia, II, III, or IV, or other formulas or compounds employed in the methods of the present invention in vivo when such prodrug is administered to a mammalian subject. Since prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc.) the compounds employed in the present methods may, if desired, be delivered in prodrug form. Thus, the present invention contemplates methods of delivering prodrugs. Prodrugs of the compounds employed in the present invention, for example, compounds of Formula I, Ia, II, III, or rv, may be prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
[0115] Accordingly, prodrugs include, for example, compounds described herein in which a hydroxy, amino, or carboxy group is bonded to any group that, when the prodrug is administered to a mammalian subject, cleaves to form a free hydroxyl, free amino, or carboxylic acid, respectively. Examples include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups; and alkyl, carbocyclic, aryl, and alkylaryl esters such as methyl, ethyl, propyl, iso-propyl, butyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, phenyl, benzyl, and phenethyl esters, and the like.
[0116] The compounds employed in the methods of the present invention may be prepared in a number of ways well known to those skilled in the art. The compounds can be synthesized, for example, by the methods described below, or variations thereon as appreciated by the skilled artisan. All processes disclosed in association with the present invention are contemplated to be practiced on any scale, including milligram, gram, multigram, kilogram, multikilogram or commercial industrial scale.
[0117] As discussed in detail above, compounds employed in the present methods may contain one or more asymmetrically substituted carbon atoms, and may be isolated in optically active or racemic forms. Thus, all chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated. It is well known in the art how to prepare and isolate such optically active forms. For example, mixtures of stereoisomers may be separated by standard techniques including, but not limited to, resolution of racemic forms, normal, reverse-phase, and chiral chromatography, preferential salt formation, recrystallization, and the like, or by chiral synthesis either from chiral starting materials or by deliberate synthesis of target chiral centers.
[0118] As will be readily understood, functional groups present may contain protecting groups during the course of synthesis. Protecting groups are known per se as chemical functional groups that can be selectively appended to and removed from functionalities, such as hydroxyl groups and carboxy groups. These groups are present in a chemical compound to render such functionality inert to chemical reaction conditions to which the compound is exposed. Any of a variety of protecting groups may be employed with the present invention. Preferred protecting groups include the benzyloxycarbonyl group and the tert-butyloxycarbonyl groups. Preferred hydroxyl protecting groups include the benzyl and the tertiary-butyldimethylsilyl groups. Other preferred protecting groups that may be employed in accordance with the present invention may be described in Greene, T.W. and Wuts, P.G.M., Protective Groups in Organic Synthesis 3rd . Ed., Wiley & Sons, 1991, or Kocienski, P. J., Protecting Groups, 3rd Ed., Georg Thieme Verlag, Stuttgart, 2005.
[0119] The compounds of the present invention are preferably combined with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice as described, for example, in Remington 's Pharmaceutical Sciences (Mack Pub. Co.,
Easton, PA, 1980), the disclosure of which is hereby incorporated herein by reference, in its entirety.
[0120] Although the compounds of the present invention may be administered as the pure chemicals, it is preferable to present the active ingredient as a pharmaceutical composition. The invention thus further provides pharmaceutical compositions comprising one or more of the cannabinoid receptor modulator compounds of the present invention, for example, compounds of Formula I, Ia, II, III, or IV, together with one or more pharmaceutically acceptable carriers therefore and, optionally, other therapeutic and/or prophylactic ingredients. The carrier(s) must be acceptable in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
[0121] In accordance with certain embodiments of the present invention, the compositions and methods of the invention may further comprise at least one cannabinoid. A variety of cannabinoids are available that may be suitable for use in the present methods and compositions. Generally speaking, it is only necessary that the cannabinoid provide the desired effect (for example, pain alleviation), and be capable of being incorporated into the present combination products and methods (discussed in detail below). In preferred embodiments, the present methods and compositions may involve a cannabinoid that is selected from Δ -tetrahydrocannabinol and cannabidiol, and mixtures thereof, more preferably, Δ9-tetrahydrocannabinol.
[0122] Alternatively, in accordance with certain embodiments of the present invention, the compositions and methods of the invention may further comprise at least one opioid. A wide variety of opioids are available that may be suitable for use in the present methods and compositions. Generally speaking, it is only necessary that the opioid provide the desired effect (for example, pain alleviation), and be capable of being incorporated into the present combination products and methods (discussed in detail below). In preferred embodiments, the present methods and compositions may involve an opioid that is selected from alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil and/or tramadol. More preferably, the opioid is selected from morphine, codeine, oxycodone, hydrocodone, dihydrocodeine, propoxyphene, fentanyl, tramadol, and mixtures thereof.
[0123] The opioid component of the present methods and compositions may further include one or more other active ingredients that may be conventionally employed in analgesic and/or cough-cold-antitussive combination products. Such conventional ingredients include, for example, aspirin, acetaminophen, phenylpropanolamine, phenylephrine, chlorpheniramine, caffeine, and/or guaifenesin. Typical or conventional ingredients that may be included in the opioid component are described, for example, in the Physicians ' Desk Reference, 1999, the disclosure of which is hereby incorporated herein by reference, in its entirety.
[0124] In addition, the opioid component may further include one or more compounds that may be designed to enhance the analgesic potency of the opioid and/or to reduce analgesic tolerance development. Such compounds include, for example, dextromethorphan or other NMDA antagonists (Mao, M. J., et al, Pain, 1996, 67, 361), L-364,718 and other CCK antagonists (Dourish, C.T., et al, Eur. J. Pharmacol, 1988, 147, 469), NOS inhibitors (Bhargava, H. N., et al., Neuropeptides, 1996, 30, 219), PKC inhibitors (Bilsky, E. J., et al, J. Pharmacol. Exp. Ther., 1996, 277, 484), and dynorphin antagonists or antisera (Nichols, M.L., et al, Pain, 1997, 69, 317). The disclosures of each of the foregoing documents are hereby incorporated herein by reference, in their entireties.
[0125] Alternatively, in accordance with certain other embodiments of the present invention, the compositions of the invention may further comprise at least one analgesic, such as for example, COX2 inhibitors, aspirin, acetaminophen, ibuprophen, naproxen, and the like, and mixtures thereof. Generally speaking, it is only necessary that the analgesic provide the desired effect (for example, pain alleviation), and be capable of being incorporated into the present combination products and methods (discussed in detail below).
[0126] In accordance with still other embodiments of the present invention, the compositions of the invention may further comprise at least one therapeutic agent selected from the group consisting of anti-seizure agents, such as, for example, carbamazepine, gabapentin, lamotrigine, and phenytoin, anti-depressants such as, for example, amitryptiline, NMDA receptor antagonists, ion channel antagonists, nicotinic receptor agonists, and anti -Parkinson's agents, such as, for example, deprenyl, amantadine, levodopa, and carbidopa. Generally speaking, it is only necessary that the anti seizure agent, anti-depressant, NMDA receptor antagonist, ion channel antagonist, nicotinic receptor agonist, or antiParkinson's agent provide the desired effect (for
example, inhibition of seizures, alleviation of depression, and the like), and be capable of being incorporated into the present combination products and methods (discussed in detail below).
[0127] The compounds of the invention may be administered in an effective amount by any of the conventional techniques well-established in the medical field. The compounds employed in the methods of the present invention including, for example, the compounds of Formula I, Ia, II, III, or IV, may be administered by any means that results in the contact of the active agents with the agents' site or site(s)of action in the body of a patient. The compounds may be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. For example, they may be administered as the sole active agents in a pharmaceutical composition, or they can be used in combination with other therapeutically active ingredients.
[0128] Compounds of the present invention can be administered to a mammalian host in a variety of forms adapted to the chosen route of administration, e.g., orally or parenterally. Parenteral administration in this respect includes administration by the following routes: intravenous, intramuscular, subcutaneous, intraocular, intrasynovial, transepithelial including transdermal, ophthalmic, sublingual and buccal; topically including ophthalmic, dermal, ocular, rectal and nasal inhalation via insufflation, aerosol and rectal systemic.
[0129] The active compound may be orally administered, for example, with an inert diluent or with an assimilable edible carrier, or it may be enclosed in hard or soft shell gelatin capsules, or it may be compressed into tablets, or it may be incorporated directly with the food of the diet. For oral therapeutic administration, the active compound may be incorporated with excipient and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like. The amount of active compound(s) in such therapeutically useful compositions is preferably such that a suitable dosage will be obtained. Preferred compositions or preparations according to the present invention may be prepared so that an oral dosage unit form contains from about 0.1 to about 1000 mg of active compound.
[0130] The tablets, troches, pills, capsules and the like may also contain one or more of the following: a binder, such as gum tragacanth, acacia, corn starch or gelatin; an excipient, such as dicalcium phosphate; a disintegrating agent, such as corn starch, potato starch, alginic acid and the like; a lubricant, such as magnesium stearate; a sweetening agent such as sucrose, lactose or
saccharin; or a flavoring agent, such as peppermint, oil of wintergreen or cherry flavoring. When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier. Various other materials may be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules may be coated with shellac, sugar or both. A syrup or elixir may contain the active compound, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavoring, such as cherry or orange flavor. Of course, any material used in preparing any dosage unit form is preferably pharmaceutically pure and substantially non-toxic in the amounts employed. In addition, the active compound may be incorporated into sustained-release preparations and formulations.
[0131] The active compound may also be administered parenterally or intraperitoneally. Solutions of the active compounds as free bases or pharmacologically acceptable salts can be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose. A dispersion can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. Under ordinary conditions of storage and use, these preparations may contain a preservative to prevent the growth of microorganisms.
[0132] The pharmaceutical forms suitable for injectable use include, for example, sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases, the form is preferably sterile and fluid to provide easy syringability. It is preferably stable under the conditions of manufacture and storage and is preferably preserved against the contaminating action of microorganisms such as bacteria and fungi. The carrier may be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol and the like), suitable mixtures thereof, and vegetable oils. The proper fluidity can be maintained, for example, by the use of a coating, such as lecithin, by the maintenance of the required particle size in the case of a dispersion, and by the use of surfactants. The prevention of the action of microorganisms may be achieved by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal and the like. In many cases, it will be preferable to include isotonic agents, for example, sugars or sodium chloride. Prolonged absorption of the injectable compositions may be achieved by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
[0133] Sterile injectable solutions may be prepared by incorporating the active compounds in the required amounts, in the appropriate solvent, with various of the other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions may be prepared by incorporating the sterilized active ingredient into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, the preferred methods of preparation may include vacuum drying and the freeze drying technique that yields a powder of the active ingredient, plus any additional desired ingredient from the previously sterile- filtered solution thereof.
[0134] The therapeutic compounds of this invention may be administered to a patient alone or in combination with a pharmaceutically acceptable carrier. As noted above, the relative proportions of active ingredient and carrier may be determined, for example, by the solubility and chemical nature of the compounds, chosen route of administration and standard pharmaceutical practice.
[0135] The dosage of the compounds of the present invention that will be most suitable for prophylaxis or treatment will vary with the form of administration, the particular compound chosen and the physiological characteristics of the particular patient under treatment. Generally, small dosages may be used initially and, if necessary, increased by small increments until the desired effect under the circumstances is reached. Generally speaking, oral administration may require higher dosages. Although the proper dosage of the products of this invention will be readily ascertainable by one skilled in the art, once armed with the present disclosure, by way of general guidance, for example, typically a daily dosage of the compound of the invention, preferably a compound as described herein, may range from about 0.001 to about 100 milligrams of the compound of the invention, preferably a compound as described herein, (and all combinations and subcombinations of ranges and specific dosage amounts therein), per kilogram of patient body weight. Preferably, the daily dosage may be about 0.01 to about 10 milligrams of the compound of the invention, preferably a compound as described herein per kilogram of patient body weight. Even more preferably, the daily dosage may be about 0.1 milligrams of the compound of the invention, preferably a compound as described herein per kilogram of patient body weight. With regard to a typical dosage form of this type, such as a tablet, the compounds of the invention, preferably a compound as described herein, generally may be present in an amount of about 0.1 to about 4 milligrams.
[0136] The combination products of this invention, such as pharmaceutical compositions comprising cannabinoids and/or opioids in combination with the compounds of Formula I, Ia, II, III, or rv, may be in any dosage form, such as those described herein, and can also be administered in various ways, as described herein. In a preferred embodiment, the combination products of the invention are formulated together, in a single dosage form (that is, combined together in one capsule, tablet, powder, or liquid, etc.). When the combination products are not formulated together in a single dosage form, the cannabinoid and/or opioid compounds and the compounds of Formula I, Ia, II, III, or IV may be administered at the same time (that is, together), or in any order. When not administered at the same time, preferably the administration of a cannabinoid and/or opioid and the compounds of Formula I, Ia, II, III, or IV occurs less than about one hour apart, more preferably less than about 30 minutes apart, even more preferably less than about 15 minutes apart, and still more preferably less than about 5 minutes apart. Preferably, administration of the combination products of the invention is oral, although other routes of administration, as described above, are contemplated to be within the scope of the present invention. Although it is preferable that the cannabinoids and/or opioids and the compounds of Formula I, Ia, II, III, or IV are both administered in the same fashion (that is, for example, both orally), if desired, they may each be administered in different fashions (that is, for example, one component of the combination product may be administered orally, and another component may be administered intravenously). The dosage of the combination products of the invention may vary depending upon various factors such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration, the age, health and weight of the recipient, the nature and extent of the symptoms, the kind of concurrent treatment, the frequency of treatment, and the effect desired.
[0137] Although the proper dosage of the combination products of this invention will be readily ascertainable by one skilled in the art, once armed with the present disclosure, by way of general guidance, where a cannabinoid and/or opioid compound is combined with the compounds of Formula I, Ia, II, III, or IV, for example, typically a daily dosage may range from about 0.01 to about 100 milligrams of the cannabinoid and/or opioid (and all combinations and subcombinations of ranges therein) and about 0.001 to about 100 milligrams of the compounds of Formula I, Ia, II, III, or IV (and all combinations and subcombinations of ranges therein), per kilogram of patient body weight. Preferably, the a daily dosage may be about 0.1 to about 10 milligrams of the cannabinoid and/or opioid and about 0.01 to about 10 milligrams of the
compounds of Formula I, Ia, II, III, or IV per kilogram of patient body weight. Even more preferably, the daily dosage may be about 1.0 milligrams of the cannabinoid and/or opioid and about 0.1 milligrams of the compounds of Formula I, Ia, II, III, or IV per kilogram of patient body weight. With regard to a typical dosage form of this type of combination product, such as a tablet, the cannabinoid compounds (e.g. Δ9-tetrahydrocannabinol or cannabidiol) and/or the opioid compounds (e.g., morphine) and generally may be present in an amount of about 15 to about 200 milligrams, and the compounds of Formula I, Ia, II, III, or IV in an amount of about 0.1 to about 4 milligrams.
[0138] Particularly when provided as a single dosage form, the potential exists for a chemical interaction between the combined active ingredients (for example, a cannabinoid and the compounds of Formula I, Ia, II, III, or IV). For this reason, the preferred dosage forms of the combination products of this invention are formulated such that although the active ingredients are combined in a single dosage form, the physical contact between the active ingredients is minimized (that is, reduced).
[0139] In order to minimize contact, one embodiment of this invention where the product is orally administered provides for a combination product wherein one active ingredient is enteric coated. By enteric coating one or more of the active ingredients, it is possible not only to minimize the contact between the combined active ingredients, but also, it is possible to control the release of one of these components in the gastrointestinal tract such that one of these components is not released in the stomach but rather is released in the intestines. Another embodiment of this invention where oral administration is desired provides for a combination product wherein one of the active ingredients is coated with a sustained-release material that effects a sustained-release throughout the gastrointestinal tract and also serves to minimize physical contact between the combined active ingredients. Furthermore, the sustained-released component can be additionally enteric coated such that the release of this component occurs only in the intestine. Still another approach would involve the formulation of a combination product in which the one component is coated with a sustained and/or enteric release polymer, and the other component is also coated with a polymer such as a low- viscosity grade of hydroxypropyl methylcellulose (HPMC) or other appropriate materials as known in the art, in order to further separate the active components. The polymer coating serves to form an additional barrier to interaction with the other component.
[0140] Dosage forms of the combination products of the present invention wherein one active ingredient is enteric coated can be in the form of tablets such that the enteric coated component and the other active ingredient are blended together and then compressed into a tablet or such that the enteric coated component is compressed into one tablet layer and the other active ingredient is compressed into an additional layer. Optionally, in order to further separate the two layers, one or more placebo layers may be present such that the placebo layer is between the layers of active ingredients. In addition, dosage forms of the present invention can be in the form of capsules wherein one active ingredient is compressed into a tablet or in the form of a plurality of microtablets, particles, granules or non-perils, which are then enteric coated. These enteric coated microtablets, particles, granules or non-perils are then placed into a capsule or compressed into a capsule along with a granulation of the other active ingredient.
[0141] These as well as other ways of minimizing contact between the components of combination products of the present invention, whether administered in a single dosage form or administered in separate forms but at the same time by the same manner, will be readily apparent to those skilled in the art, once armed with the present disclosure.
[0142] It will be further appreciated that the amount of the compound, or an active salt or derivative thereof, required for use in treatment will vary not only with the particular salt selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will be ultimately at the discretion of the attendant physician or clinician.
[0143] The desired dose may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day. The sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations; such as multiple inhalations from an insufflator or by application of a plurality of drops into the eye.
[0144] The dose may also be provided by controlled release of the compound, by techniques well known to those in the art.
[0145] Pharmaceutical kits useful in, for example, the treatment of pain, which comprise a therapeutically effective amount of a cannabinoid and/or opioid along with a therapeutically
effective amount of a pyridine compound of the invention, in one or more sterile containers, are also within the ambit of the present invention. Sterilization of the container may be carried out using conventional sterilization methodology well known to those skilled in the art. The sterile containers of materials may comprise separate containers, or one or more multi-part containers, as exemplified by the UNIVIAL™ two-part container (available from Abbott Labs, Chicago, Illinois), as desired. The opioid or cannabinoid compound and the compound of Formula I, II, III, or IV may be separate, or combined into a single dosage form as described above. Such kits may further include, if desired, one or more of various conventional pharmaceutical kit components, such as for example, one or more pharmaceutically acceptable carriers, additional vials for mixing the components, etc., as will be readily apparent to those skilled in the art. Instructions, either as inserts or as labels, indicating quantities of the components to be administered, guidelines for administration, and/or guidelines for mixing the components, may also be included in the kit.
[0146] In certain preferred embodiments, the compounds, pharmaceutical compositions and methods of the present invention may involve a peripheral cannabinoid receptor agonist compound. The term "peripheral" designates that the compound acts primarily on physiological systems and components external to the central nervous system. In preferred form, the peripheral receptor agonist compounds employed in the methods of the present invention exhibit high levels of activity with respect to peripheral tissue, such as, gastrointestinal tissue, while exhibiting reduced, and preferably substantially no CNS activity. The phrase "substantially no CNS activity," as used herein, means that less than about 20% of the pharmacological activity of the compounds employed in the present methods is exhibited in the CNS, preferably less than about 15%, more preferably less than about 10%, even more preferably less than about 5%, and most preferably 0%, of the pharmacological activity of the compounds employed in the present methods is exhibited in the CNS.
[0147] The compounds of the present invention may be used in methods to bind cannabinoid receptors, more preferably CB1 or CB2 cannabinoid receptors. Such binding may be accomplished by contacting the receptor with an effective amount of a compound of Formula I, II, III, or IV. The cannabinoid receptors may be located in the central nervous system or located peripherally to the central nervous system or in both locations. Preferably, the contacting step conducted in an aqueous medium, preferably at physiologically relevant ionic strength, pH, and the like.
[0148] Furthermore, it is preferred in certain embodiments of the invention where the compound is administered to agonize the peripheral cannabinoid receptors that the compound of the invention administered does not substantially cross the blood-brain barrier and thereby reduces the classical central side effects as observed for blood-brain penetrating cannabinoid agonists such as Δ9-tetrahydrocannabinol (Δ9-THC). The central side effects of blood brain penetrating cannabinoid agonists limits their clinical utility, such as their use in the relief of pain. The phrase "does not substantially cross," as used herein, means that less than about 30% by weight of the compound employed in the present methods crosses the blood-brain barrier, preferably less than about 15% by weight, more preferably less than about 10% by weight, even more preferable less than about 5% by weight and most preferably 0% by weight of the compound crosses the blood-brain barrier. Selected compounds can be evaluated for CNS penetration by determining plasma and brain levels following i.v. administration.
[0149] In yet another aspect, the invention is directed to methods of binding cannabinoid receptors, preferably CB1 and/or CB2 receptors, comprising the step of administering to a patient in need thereof, an effective amount of a compound of the invention including, for example, a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
[0150] In certain preferred aspects, the methods comprise the step of administering to said patient an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
[0151] In some preferred embodiments, the cannabinoid receptors are CB1 and/or CB2 cannabinoid receptors. In certain more preferred embodiments, the compound selectively binds CB2 cannabinoid receptors relative to CB1 receptors, even more preferably peripheral CB2 receptors. In certain preferred embodiments, the cannabinoid receptors are located in the central nervous system. In other preferred embodiments, the cannabinoid receptors are located peripherally to the central nervous system. In some other preferred embodiments, the compound exhibits activity toward the cannabinoid receptors. In certain preferred embodiments, the binding agonizes the activity of the cannabinoid receptors. In other preferred embodiments, the binding antagonizes the activity of the cannabinoid receptors. In still other preferred embodiments, the binding inversely agonizes the activity of the cannabinoid receptors.
[0152] In certain embodiments, the present invention is directed to methods of treating a gastrointestinal disorder, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or FV, or any combination thereof.
[0153] In certain preferred embodiments, the gastrointestinal disorders which may be treated with the present compounds and methods include, for example, nausea, vomiting, loss of appetite, cachexia, diarrhoea, inflammatory bowel disease, or irritable bowel syndrome, or any combination thereof.
[0154] In some embodiments, the present invention is directed to methods of treating inflammation, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
[0155] In certain embodiments, the present invention is directed to methods of treating autoimmune diseases, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or FV, or any combination thereof.
[0156] In some preferred embodiments, the autoimmune disease is multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, systemic lupus erythematosus, myasthenia gravis, diabetes mellitus type I, osteoporosis, or any combination thereof.
[0157] In some embodiments, the present invention is directed to methods of treating an immune related disorder, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
[0158] In certain preferred embodiments, the immune related disorder is asthma, chronic pulmonary obstructive disorder, emphysema, bronchitis, allergy, tissue rejection in organ transplants, celiac disease, or Sjogren's syndrome, or any combination thereof.
[0159] In certain embodiments, the present invention is directed to methods of treating pain, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or FV, or any combination thereof.
[0160] In embodiments involving the treatment of pain, the pain may be inflammatory pain, neuropathic pain, visceral pain, surgical pain, including pain which occurs during surgery or pain which occurs after surgery (i.e., postsurgical pain), or cancer related pain. In certain more preferred embodiments, the present pain ameliorating methods may further comprise the administration to the patient of at least one opioid in the form of combination products and/or combination therapy. Suitable opioids include, for example, alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil or tramadol, and mixtures thereof. In embodiments involving the treatment or prevention of neuropathic pain, the present methods may further comprise administering to a patient codeine, carbamazepine, gabapentin, lamotrigine, phenytoin, amitryptiline, an NMDA receptor antagonist, an ion channel antagonist, or a nicotinic receptor agonist, or a mixture thereof, in the form of combination products and/or combination therapy.
[0161] In some preferred embodiments, the methods for treating pain may further comprise the administration to the patient of at least one cannabinoid.
[0162] In some embodiments, the present invention is directed to methods of treating hypertension, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
[0163] In some embodiments, the present invention is directed to methods of treating neurodegenerative diseases, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
[0164] In some preferred embodiments, the neurodegenerative disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, or amyotrophic lateral sclerosis. In certain more preferred embodiments, these methods may further comprise the administration to the patient of deprenyl, amantadine, levodopa, or carbidopa, in the form of combination products and/or combination therapy.
[0165] In other embodiments, the present invention is directed to methods of treating neurological disorders, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or FV, or any combination thereof.
[0166] In certain preferred embodiments, the neurological disorder is stroke, migraine, or cluster headache, or any combination thereof.
[0167] In other embodiments, the present invention is directed to methods of providing cardioprotection against ischemic and reperfusion effects, comprising the step of administering to a patient in need thereof, an effective amount of a compound of formula I, Ia, II, III, or IV, or any combination thereof.
[0168] In certain preferred embodiments, the ischemic or reperfusion effect is arrhythmia or hypertension, or a combination thereof.
[0169] In some embodiments, the present invention is directed to methods of inhibiting mechanical hyperalgesia associated with nerve injury, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof.
[0170] In other embodiments, the present invention is directed to methods of inducing apoptosis in malignant cells, comprising the step of contacting said cells with an effective amount of a compound of Formula I, Ia, II, III, or FV, or any combination thereof.
[0171] In some preferred embodiments, the apoptosis occurs in vitro, In other preferred embodiments, the apoptosis occurs in vivo.
[0172] In other embodiments, the present invention is directed to methods for modulating appetite, comprising the step of administering to a patient in need thereof, an effective amount of a compound of Formula I, Ia, II, III, or IV, or any combination thereof. In some preferred embodiments, the modulating decreases appetite. In other preferred embodiments, the modulating enhances appetite.
METHODS OF PREPARATION
[0173] Employing the methodology herein described or cited, pyridine compounds of Formula I, Ia, II, III, or IV can be readily prepared. The invention is further described in the following examples. The actual examples, herein provided, are for illustrative purposes only, and are not to be construed as limiting the appended claims. They provide a series of pyridine derivatives (IA-I ID) of Formula I, Ia, II, III, or IV, prepared according to Schemes 1-25, shown below in Examples 1 A-25G. Likewise, The prophetic examples 12, 13, and any intermediates thereto, herein provided in Schemes 12 and 13, are for illustrative purposes only, and are not to be construed as limiting the appended claims.
[0174] Compound IA was prepared as outlined in Scheme 1. Commercially available (5- aminopyridin-3-yl)-methanol 1.1 was reacted with acid chloride al and the resulting ester amide was treated with ethanol/NaOHaq to obtain 7V-(5-(hydroxymethyl)pyridin-3-yl)-2- methylcyclohexane-carboxamide 1.2. Reaction of 1.2 with mesyl chloride followed by treatment with morpholine yielded 2-methyl-N-(5-(morpholinomethyl)pyridin-3- yl)cyclohexanecarboxamide IA.
[0175] Compounds 2A-2V were prepared as outlined in Scheme 2. Commercially available 5- bromo-4-methylpyridin-2-amine 2.1 was treated with acid chlorides a2, a3, a5 or a6 to yield amides 2.2a2, 2.2a3, 2.2a5 or 2.2a6, respectively. Bromination of 2.2a2, 2.2a3, 2.2a5 or 2.2a6 using NBS/AIBN followed by reaction with amines bl-b9 gave compounds 2A, 2F, 2H, 2M, 2P, 2T-2V or intermediates 2.4a3bl, 2.4a2b4, 2.4a5b4, 2.4a6b4, 2.4a2b5, and 2.4a2b6. Replacement of the 4-bromo substituent in 2 A, 2F, 2H, or intermediate 2.4a3bl with methylzinc chloride cl, boronic acids c2, c3, or c4 generated compounds 2B-2E, 2G, or 21. Reaction of 2 A with sodium methoxide in methanol c5, produced compound 2 J. Reaction of 2.4a3bl with 2- methoxyethanamine c6, yielded compound 2K. Replacement of the 4-bromo substituent in 2M, 2P or intermediates 2.4a2b4, 2.4a5b4, 2.4a6b4, 2.4a2b5, and 2.4a2b6 with methylzinc chloride cl, generated compounds 2L, 2N, 2O, 2Q and 2R. Compound 2S was obtained from 2 A via a palladium catalyzed CO insertion followed by an ester cleavage and a Curtius reaction.
[0176] Compounds 3A-3O were prepared as outlined in Scheme 3. Hydrolysis of compound 2B under acidic conditions gave intermediate 3.1. Reaction of 3.1 with acid chlorides al, a4, a7, a8, or al0-al3 produced compounds 3 A - 3H respectively. Reaction of 3.1, the coupling
reagent Bop-Cl (bis(2-oxo-3-oxazolidinyl)phosphinic chloride) and acids el - e7 produced compounds 31 - 3O.
[0177] Compound 4A was prepared as outlined in Scheme 4. Treatment of 3.1 with isocyanate 4.1 gave urea 4A.
[0178] Compounds 5A-5E were prepared as outlined in Scheme 5. Commercially available 5- bromo-4,6-dimethylpyridin-2-amine 5.1 was coupled with acid chloride a2 to give intermediate 5.2. Bromination of 5.2 using NBS/ AIBN generated regioisomers 5.3 and 5.4. Treatment of compounds 5.3 and 5.4 with bl gave 5 A and 5B respectively. Reaction of compounds 5A and 5B with cl yielded compounds 5C and 5E respectively. Compound 5D was also isolated during the synthesis of compound 5C.
[0179] Compounds 6A-6B were prepared as outlined in Scheme 6. Treatment of commercially available 6-amino-3-bromo-2-methylpyridine with acid chloride a2 generated intermediate 6.2. Bromination of 6.2 using NBS/AIBN produced compound 6.3, which was converted to compound 6 A using amine bl. Reaction of 6A with cl produced compound 6B.
[0180] Compound 7A was prepared as outlined in Scheme 7. Conversion of commercially available 4,6-dimethylpyridin-2-amine 7.1 to amide 7.2 was achieved by treatment with acid chloride a2. Bromination of 7.2 using NBS/AIBN produced compound 7.3 which was converted to compound 7A using amine bl.
[0181] Compounds 8A-8B were prepared as outlined in Scheme 8. 4-Hydroxyquinolin-2- (1H)-one 8.1 was converted to dichloride 8.2 using phosphorous oxychloride. Reaction of 8.2 with amine bl gave intermediate 8.3. Treatment of 8.3 with ammonium hydroxide gave 8.4 which was then reacted with acid chlorides a2 and al to generate compounds 8A and 8B respectively.
[0182] Compound 9 A was prepared as outlined in Scheme 9. Using a literature procedure described by Otsubo (Chemical & Pharmaceutical Bulletin 1991, 39(11), 2906-9) 4- (Bromomethyl)quinolin-2(1H)-one 9.1 was converted to 9.2 using phosphorous oxychloride. Treatment of 9.2 with amine bl gave intermediate 9.3, which generated 9.4 after reaction with
aqueous ammonia at elevated temperature and pressure. Reaction of amine 9.4 with acid chloride a2 gave compound 9 A.
[0183] Compounds 10A-10O were prepared as outlined in Scheme 10. Condensation of ketones dl, d2 or d3 with diethyl oxalate 10.1 and 2-cyanoacetamide 10.2 followed by acid hydrolysis in concentrated hydrochloric acid yielded acids 10.3dl, 10.3d2 or 10.3d3 respectively. Chlorinated compounds 10.4dl, 10.4d2 or 10.4d3 were generated by treatment of lactams 10.3dl, 10.3d2 or 10.3d3 with phosphorus oxychloride. Reaction of acid chlorides 10.4dl, 10.4d2 or 10.4d3 with amine bl or b6 generated amides 10.5dlbl, 10.5d2bl, 10.5d3bl or 10.5dlb6, which were reduced to their respective amines 10.6dlbl, 10.6d2bl, 10.6d3bl or 10.6dlb6 using borane-methyl sulfide complex. Displacement of the 2-chloro substituent with hydrazine followed by reduction of the hydrazine moiety gave intermediates 10.7dlbl, 10.7d2bl, 10.7d3bl or 10.7dlb6. Condensation of these amines with acid chloride a2 yielded compounds 10A, 10B or 10C. Condensation of 10.7dlbl with acid chlorides a8, a9, al3, all, a4 or a6 gave compounds 10D - 101 respectively. Condensation of 10.7dlb6 with acid chlorides a2, a9 or all yielded compounds 10J - 10 L. Reaction of chloride 10.6dlbl with cycloheptylamine, 1-methoxypropan-2-amine or (i/?,2i?,^5)-bicyclo[2.2.1]heptan-2-amine yielded compounds 10M, 10N and 10O respectively.
[0184] Compounds 11 A-IlD were prepared as outlined in Scheme 11. Reduction of compounds IA, 10A, 2B and 10C with borane-methyl sulfide complex gave compounds 11A-D.
[0185] Schemes 12 and 13 are prophetic. Commercially available 5,6,7,8- tetrahydroisoquinoline 12.1 is oxidized with hydrogen peroxide (Gribble et al., Tetrahedron 1988, 44(11), 3195-3202) and chlorinated with phosphorous oxychloride (Yamanaka et al., Chemical & Pharmaceutical Bulletin 1988, 36(6), 2244-7) to yield 12.2. Conversion to 12.3 is accomplished following a procedure described by Mac Bride et al., Synthetic Commun. 1996, 2(5(12), 2309-2316. A Wittig reaction followed by hydroboration applicable to the synthesis of 12.5 is described in WO20040052851. Synthesis of aldehyde 12.6 is achieved through Swern oxidation (Mancuso and Swern, Synthesis 1981, 165-185). Reductive animation with morpholine and sodium cyanoborohydride leads to 12.7 followed by conversion of the chloro- substituent to amine 12.8 according to example 8. Amide 12 is obtained following general method A (Scheme 12).
[0186] Previously synthesized chloropyridines 10.6a-c is reacted with copper cyanide and a palladium catalyst to produce cyanopyridine 13.1, which is hydrolyzed to acid 13.2 with hydrochloric acid. Acid 13.2 is converted to the acid chloride with thionyl chloride and coupled with an amine to yield amide 13.
[0187] Compounds 14A-14AG were prepared as outlined in Scheme 14. Replacement of the 4-bromo substituent in 2 A with boronates c7or c8 generated compounds 14.1c7 or 14.1c8, which were hydrogenated to give compounds 14A and 14B respectively. The amide in 14B was cleaved under acidic conditions and then reacted with acid chlorides a4, a10, a6, al4, all, a5, a8, Ά9 or al3 to give compounds 14C - 14K.
[0188] Compound 2 A was reacted with sodium methoxide c9 and copper bromide to yield amine 14.2c9, which was reacted with Bop-Cl and acids e8, e4, e9, el, e10, e5, ell or with acid chloride a2 to yield compounds 14L - 14S respectively.
[0189] Compound 2A was reacted with sodium ethoxide c10 and copper bromide to yield amine 14.2c10, which was reacted with acid chlorides or acids (acids employed with BOP added) a2, a4, a10, al4, e9, el, e2, a6, a8, e5, or ell to yield compounds 14T - 14AD respectively.
[0190] Compounds 14AE - 14AG were prepared using methane thiolate c10, ethane thiolate ell and acid chlorides a3, al4 or a2 respectively.
[0191] Compound 15A was prepared as outlined in Scheme 15. Commercially available methyl 2-amino-6-methylisonicotinate 15.1 was brominated, converted to amide 15.3 and reduced to 15.4 with borane dimethyl sulfide complex. The 4-bromo substituent in 15.4 was replaced through a reaction with sodium methoxide and copper bromide to give 15.5, followed by amide formation with acid chloride a2 to yield compound 15 A.
[0192] Compound 16A was prepared as outlined in Scheme 16. Commercially available 3- (methoxymethoxy)pyridine 16.1 was reacted with n-butyl lithium, DMF and TMEDA to form aldehyde 16.2. Reductive amination with bl yielded intermediate 16.3. Deprotection with HCl in ether produced 16.4 which was reacted with sodium iodide to yield 16.5. Alkylation to 16.6 was accomplished with 1-iodopropane and potassium carbonate. Intermediate 16.7 was
obtained through a palladium catalyzed reaction with tert-butyl carbamate. Compound 16A was prepared from 16.7, acid el and a coupling reagent (Bop-Cl).
[0193] Compounds 17A - 17AA were prepared as outlined in Scheme 17. Methyl 2-(2,2- dimethylbutanamido)-5-methylisonicotinate (17.1, obtained from methyl 2-amino-5- bromoisonicotinate and reactions according to general methods A and D) was reduced to alcohol 17.2 with DiBAL and oxidized to aldehyde 17.3 with pyridinium chlorochromate. Compounds 17A - 17AB were obtained in a parallel fashion through reductive amination with amines f1 - f25 and polymer bound cyanoborohydride. Grignard reaction of 17.3 with ethylmagnesium bromide yielded alcohol 17.4, which was converted to the bromide with carbon tetrabromide and reacted with amine b1 to yield compound 17AA.
[0194] Compounds 18A - 18D were prepared as outlined in Scheme 18. Commercially available 4-chloro-2,3-dimethylpyridine-7V-oxide 18.1 was reacted with potassium tert-butoxide and benzyl alcohol and then chlorinated with POCl3. Chloride 18.3 was reacted with 2,2- dimethylbutanamide under palladium catalysis, then deprotected and converted to the triflate 18.6. This intermediate was reacted with 4-fluorophenethylzinc bromide, pyridylzinc bromide or benzylzinc bromide to yield compounds 18 A, 18B or 18C respectively. Triflate 18.6 was converted to ester 18.7 via a palladium catalyzed CO insertion reaction. Ester reduction, bromination and reaction with amine b4 yielded compound 18D.
[0195] Compounds 19A and 19B were prepared as outlined in Scheme 19. Commercially available 4-bromopyridin-2-amine 19.1 was coupled with phenyl boronic acid under Suzuki conditions and brominated with bromine in chloroform. Intermediate 19.3 was coupled with acid chloride a2 to yield compound 19 A. Intermediate 19.3 was reacted with sodium methoxide under copper catalysis and then coupled with acid chloride a2 to yield compound 19B.
[0196] Compound 20A was prepared as outlined in Scheme 20. Intermediate 10.3dl was chlorinated with POCl3, esterified with methanol and HCl in dioxane and then coupled with 2- cyclopropylisobutyramide under palladium catalysis to yield intermediate 20.2. Reduction with DIBAL, bromination and reaction with amine b4 produced compound 20A.
[0197] Compound 21 A - 21D were prepared as outlined in Scheme 21. Intermediate 2.3a2 was reacted with triethyl phosphate. The phosphonate 21.1 was reacted under Horner
Wadsworth Emmons conditions with 1-acetylpiperidin-4-one or dihydro-2H-pyran-4(3H)-one. Intermediate 21.2a or 21.2c was reacted with methyl zinc chloride and reduced to compounds 21 A or 21C. Intermediate 20.4 was reacted with triethyl phosphate, coupled with 1- acetylpiperidin-4-one and reduced to compound 21B.
Intermediate 21.1 was coupled with dihydro-2H-thiopyran-4(3H)-one and oxidized to sulfone 21.4 with peroxyacetic acid. Reaction with methyl zinc chloride, followed by reduction with H2/Pd/C yielded compound 21D.
[0198] Compound 22 A was prepared as outlined in Scheme 22. Commercially available 2- (dimethylammomethyl)-3-hydroxypyridine (22.1) was methylated and converted to 22.3 with trimethylsulfbxonium iodide and sodium hydride. Dihydrofuropyridine 22.3 was nitrated , reduced and coupled to acid chloride a2. Intermediate 22.6 was oxidized with mCPBA, brominated and coupled to c8 under Suzuki conditions. Treatment with osmium tetroxide and sodium iodate yielded aldehyde 22.10. Reductive amination with bl followed by reduction with Raney nickel yielded compound 22A.
[0199] Compound 23A was prepared as outlined in Scheme 23. Intermediate 2.3a2 was reacted with potassium cyanide and coupled with methyl boronic acid under Suzuki conditions. The cyanide 23.2 was reduced under rhodium catalysis and reacted with 1-bromo-2-(2- bromoethoxy)-ethane and potassium carbonate to form compound 23A.
[0200] Compounds 24A - 24G were prepared as outlined in Scheme 24. 3-Methyl-4- nitropyridine 1-oxide (24.1a), 2,3-dimethyl-4-nitropyridine 1-oxide (24.1b) or 4-nitro-6,7- dihydro-5H-cyclopenta[b]pyridine 1-oxide (24.1c) were reacted with acetyl chloride and sodium sulfide, followed by treatment with chlorine and 9M HCl and reaction with bl . Intermediate 24.4 was chlorinated with POCl3 and coupled to 2,2-dimethylbutan-amide or 2-cyclopropyl-2- methylpropanamide to produce compounds 24A - 24F. Intermediate 24.5 was coupled to cyclohexyl amine under palladium catalysis to yield compound 24G.
[0201] Compounds 25A - 25G were prepared as outlined in Scheme 25. Intermediate 2.3a2 was reacted with methylamine and then coupled with 4-fluorophenylsulfonylchloride or 4- trifluoromethylphenylsulfonylchloride to produce compounds 25A or 25B respectively. Reaction of 2.3a2 with methylbenzyl amine, followed by replacement of the bromo-substituent through reaction with methyl zinc chloride, amide cleavage and coupling with acid chloride a6
yielded compound 25C. Debenzylation of 25C with H2/Pd/C and coupling of the resulting amine 25.3 with 4-fluorophenylsulfonylchloride or 4-trifluoromethylphenylsulfonylchloride gave compounds 25D and 25E. Reaction of 2.3a2 with Boc-piperazine, reaction with methyl zinc chloride, deprotection and coupling with 6-isocyanato-2,3-dihydrobenzo[b][1,4]dioxine produced compound 25F. Reaction of 2.3a2 with benzylpiperazine, followed by reaction with methyl zinc chloride, amide cleavage, coupling with acid chloride a6, deprotection and coupling with 1,2,3-thiadiazole-4-carbonyl chloride yielded compound 25G.
EXPERIMENTAL PROCEDURES
[0202] Materials: All chemicals were reagent grade and used without further purification. LC-MS data were obtained using a Thermo-Finnigan Surveyor HPLC and a Thermo-Finnigan AQA MS using positive or negative electrospray ionization. Program (positive) Solvent A: 10 mM ammonium acetate, pH 4.5, 1% acetonitrile; solvent B: acetonitrile; column: Phenomenex LUNA C18(2), 30 x 2.00 mm, 3μ, detector: PDA λ = 220-300 nm. Gradient: 96%A-100%B in 3.2 minutes, hold 100%B for 0.4 minutes. Program (negative) 1 mM ammonium acetate, pH 4.5, 1% acetonitrile; solvent B: acetonitrile; column: Phenomenex LUNA C18(2), 30 x 2.00 mm, 3μ, detector: PDA λ = 220-300nm. Gradient: 96%A-100%B in 3.2 minutes, hold 100%B for 0.4 minutes.
Abbreviations used in these examples
ACN acetonitrile
AIBN 2,2' -azobisisobutyronitrile
CCl4 carbon tetrachloride
DCM dichloromethane
DIEA diisopropyl ethyl amine
DMF dimethyl formamide
EtOAc ethyl acetate
NBS N-bromo-succinimide
TEA triethylamine
Common Reagents used during the synthesis of examples 1-25G:


Scheme 2:

Scheme 3:

3.

Scheme 5:
5.3a-b
5E


Scheme 7:
7.3

Scheme 8:
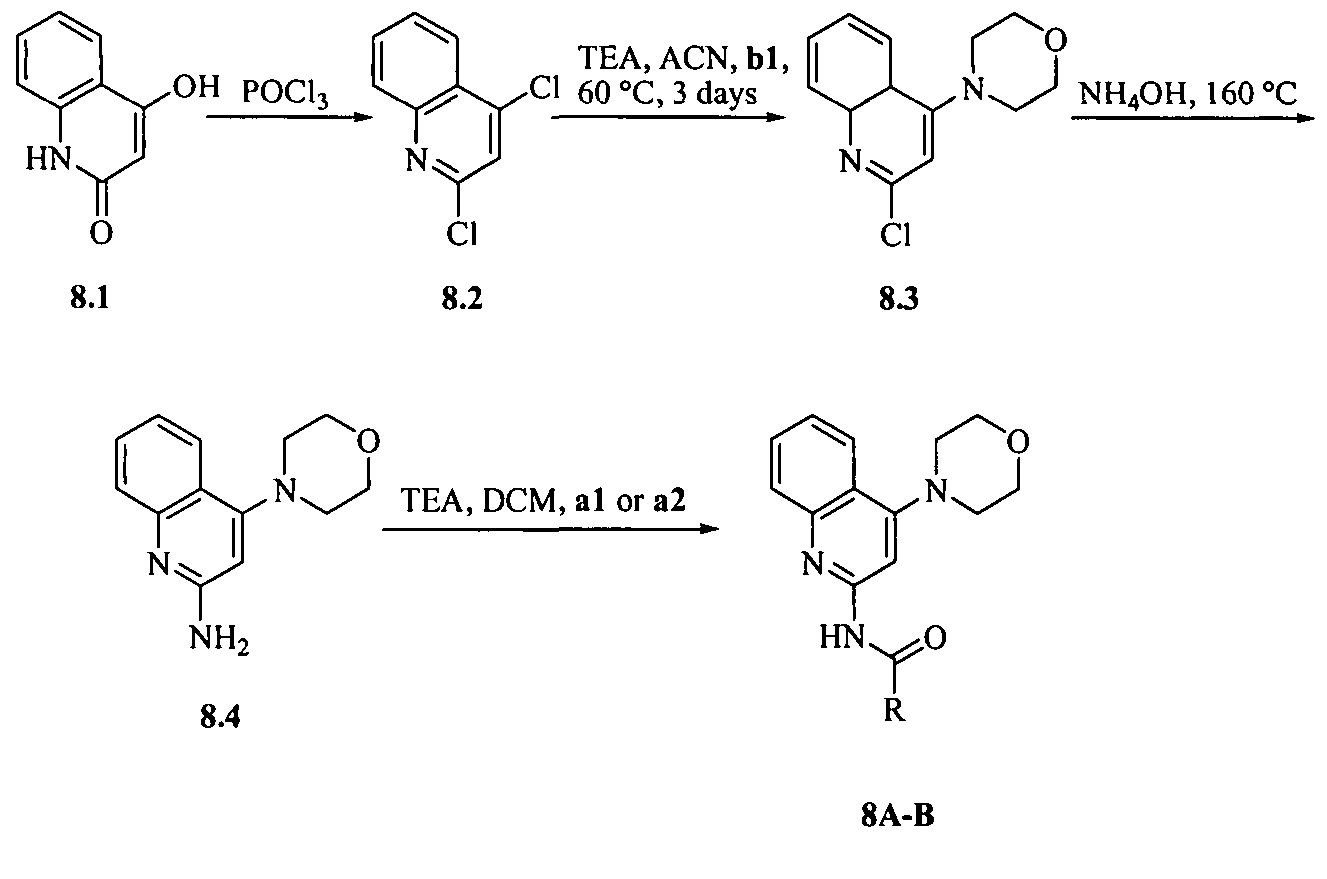

Scheme 10:
10. 10.

Scheme 11:

-70-
Scheme 12:
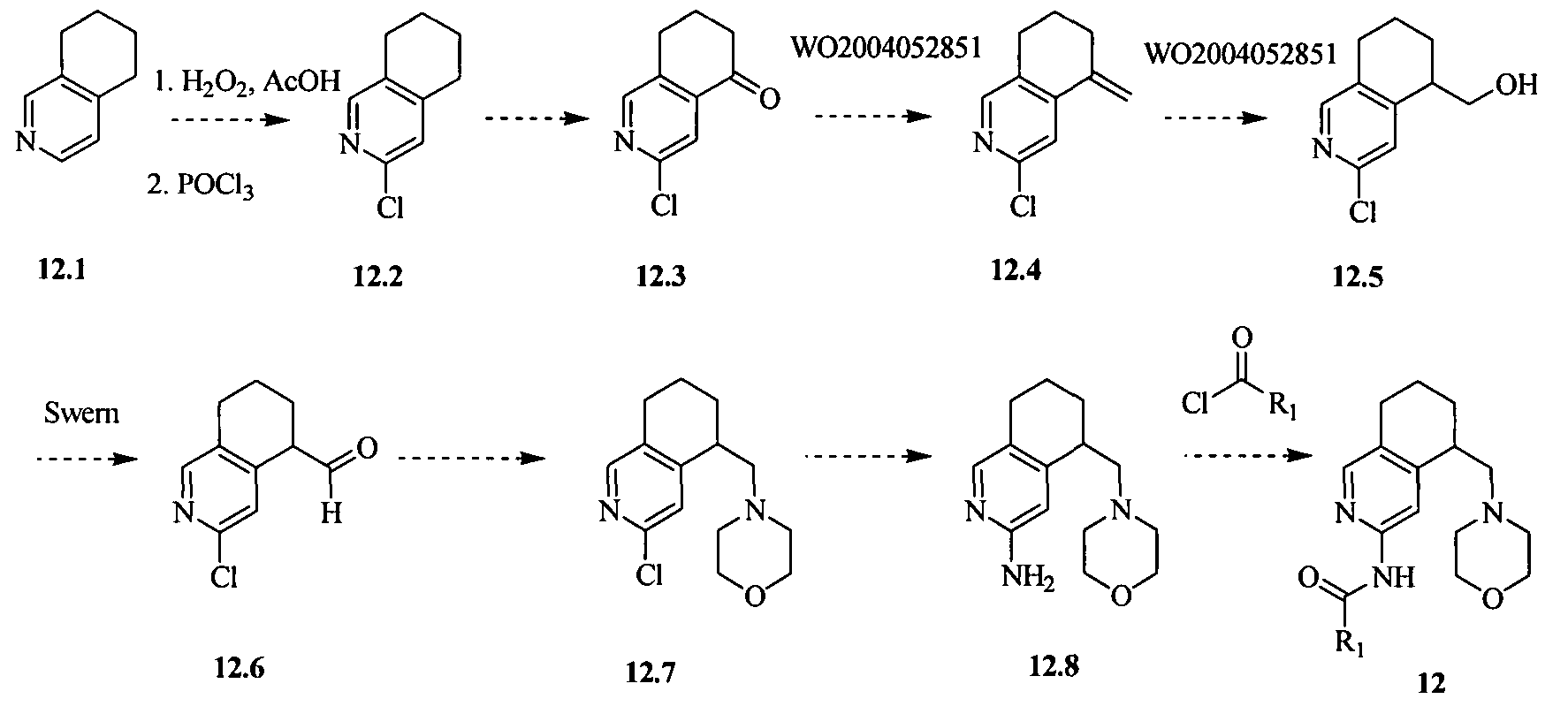
Scheme 13

Scheme 14


Scheme 16
16.1 16.3 16.4

Scheme 17
17ΛA
Amines used to

Scheme 18
18.7 18.9 18D

Scheme 19

20.2 20.4

Scheme 21

Scheme 22

Scheme 23


Scheme 25
25A, B

GENERAL METHODS:
A. Preparation of Amides
[0203] To amine (e.g. 2.1, 10.16 g, 5.432 mmol) in DCM (130 mL) was added NJt- diisopropyl-ethylamine (10.9 mL, 6.25 mmol). The reaction was cooled to 0 °C and a solution of acid chloride (e.g. a2, 7.83 mL, 5.70 mmol) was added slowly. The reaction was stirred overnight at room temperature. The reaction was washed with 1 N HCl, saturated NaHCO3 solution, and brine, dried over MgSO4, filtered, the solvent evaporated and the residue was purified by silica gel chromatography using hexane/ethyl acetate eluents.
B. Bromination with NBS/AIBN
[0204] To a solution of methylpyridine carboxamide (e.g. 2.2, 1.72 g, 6.0 mmol) in CCl4 (30 mL) was added NBS (1.40 g, 7.84 mmol) followed by AIBN (0.0990 g, 0.603 mmol). The reaction mixture was refluxed for 4 hours. (If the reaction was not complete, then another 0.3 eq. of NBS (321 mg, 1.8 mmol) and 0.1 eq. of AIBN (99 mg) were added and refluxed overnight.). The reaction was cooled to RT and filtered through a Celite pad. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography using EtOAc- hexane (1:8) as eluents.
C. Preparation of Benzamines
[0205] The brominated product (e.g. 2.3, 1.70 g, 4.67 mmol) was dissolved in THF (25 mL), NaHCO3 (630 mg, 7.5 mmol) was added followed by addition of amine (e.g. bl, 2.1 mL, 24 mmol). The reaction mixture was stirred at room temperature for two days. The reaction mixture was diluted with EtOAc and washed with water. The organic layer was dried and concentrated. The residue was purified by column chromatography using acetone-hexane (1:10) as eluent.
D. Reaction with Methylzinc Chloride
[0206] To a solution of benzamine amide (e.g. 2A, 0.400 g, 1.08 mol) in THF (20 mL) at room temperature was added tetrakis(triphenylphosphine)palladium(0) (0.125 g, 0.108 mmol) followed by dropwise addition of 2.0 M of methylzinc chloride cl, in tetrahydrofuran (2.7 mL). The reaction was stirred at 50-55 °C overnight. The reaction was quenched with saturated NH4Cl and extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was evaporated and the residue was dissolved in ether, extracted with 1 N HCl. The aqueous layer was basified with aqueous NaOH, and extracted with DCM. The organic layers were combined, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using acetone-hexane (1:2) as eluent.
E. Suzuki Coupling
[0207] A solution of aryl bromide (e.g. 2A, 200 mg, 0.5 mmol), boronic acid (e.g. c2, 64.5 mg, 0.525 mmol), potassium carbonate (198 mg, 1.43 mmol) and tetrakis(triphenylphosphine)palladium(0) (55 mg, 0.048 mmol) in water (0.9 mL) and 1,4-
dioxane (3 mL) was heated in the microwave at 150 °C for 2 hours. The mixture was filtered through celite and the solvents evaporated. The residue was dissolved in a very small amount of DCM. 1.0 M HCl and ether were then added. The organic layer was separated and the aqueous layer was basified with sodium bicarbonate. DCM was added and the organic layer was dried with magnesium sulfate and the solvent was evaporated. The crude product was purified by preparative HPLC.
Acid Salts
[0208] Compounds IA, 2F, 2H, 2K, 3B, 3D, 31 and 8A were prepared as HCl salts (by dissolving the compound in HCl in ether or dioxane and evaporating the solvents, similar as in example IA). Only free-base structures were used in the schemes provided
EXAMPLE l:
Preparation of N-^-hydroxymethyl)pyridin^-yl)-l-methylcyclohexanecarboxamide (1.2)

[0209] A 20 mL screw top vial with a septa was charged with 2-amino-4- hydroxymethylpyridine (1.1, 902 mg, 0.00726 mol), methylene chloride (8 mL, 0.1 mol) and N,N-diisopropyl-ethylamine (3160 μL, 0.0182 mol). The reaction was cooled in an ice/water bath and the 2-methylcyclohexanecarbonyl chloride (2570 mg, 0.0160 mol) was added drop wise over ten minutes and the reaction was allowed to slowly warm to room temperature and stir for 30 hours. The reaction was diluted with 20 mL of water, 20 mL of DCM and 20 mL of ethyl acetate. The phases were separated and the aqueous phase was back extracted with 30 mL of ethyl acetate. The combined organic phases were concentrated under vacuum and redissolved in 20 mL of ethanol. Then 4 mL of 1.0 N NaOH was added, followed by the addition of 1.6 grams of solid sodium hydroxide. After 90 minutes the reaction was concentrated under vacuum and partitioned between 70 mL of ethyl acetate and 30 mL of water. The aqueous phase was back extracted with 40 mL of ethyl acetate. The combined organic extracts were concentrated under vacuum to yield 1.4 g of 1.2 as oil (yield 71%).
MS analysis m/τ = 248+1.
Preparation of 2-methyl-N-(4-(morpholinomethyl)pyridinyl-2-yl)cyclohexane carboxamide (IA)

[0210] A 10 mL flask was charged with 7V-(4-hydroxymethyl)pyridin-2-yl)-2-methylcyclo- hexanecarboxamide (1.2, 290 mg, 0.0012 mol) and methylene chloride (6 mL, 0.09 mol). The solution was cooled in an ice/water bath and the methanesulfonyl chloride (104 μL, 0.00134 mol) was added at once. After a 10 minutes in the cooling bath N,7V-diiso-propylethylamine (260 μL, 0.0015 mol) was added at once to the ice cooled reaction and after 1 hour morpholine (320 μL, 0.0037 mol) was added at once and the reaction was allowed to slowly warm to room temperature and stir overnight. The reaction was diluted with 35 mL of ethyl acetate and washed with 25 mL of water. The aqueous phase was back extracted with 25 mL of ethyl acetate and the combined extracts were dried with sodium sulfate and concentrated under vacuum. The material was chromatographed on a 12 gram silica gel column with a gradient of 15 to 50 % ethyl acetate/hexanes. The oil obtained was dissolved in 4 mL of methanol and 1.5 mL of HCl in dioxane was added. After ten minutes the solvents were removed under vacuum and the sample dried overnight. The yield was 123 mg (32%).
1H NMR (CD3OD), δ 1.00 (d, J= 7 Hz, 3H), 1.44 (m, 2H), 1.66 (m, 2H), 1.77 (m, 2H), 2.24 (br s, 1H), 2.85 (m, 1H), 3.24 (t, J= 5 Hz, 1H), 3.45 (m, 4H), 3.90 (m, 1H), 4.02 (br s, 4H), 4.67 (s, 2H), 7.89 (dd, J= 6 Hz and 2 Hz, 1H), 7.93 (s, 1H), 8.45 (d, 1H). MS analysis m/z = 317+1.
EXAMPLE 2A;
Preparation of N-(5-bromo-4-methylpyridin-2-yl)-2,2-dimethylbutanamide (2.2)
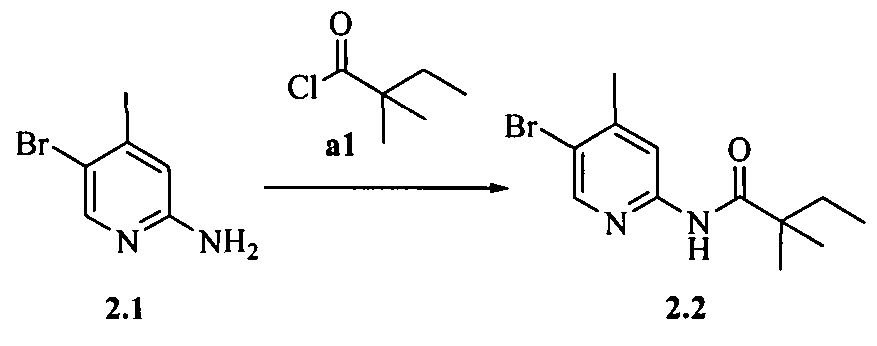
Preparation of N-(5-bromo-4-(bromomethyl)pyridin-2-yl)-2,2-dimethylbutanamide (2.3)
2.3

[0212] 7V-(5-bromo-4-(bromomethyl)pyridin-2-yl)-2,2-dimethylbutanamide 2.3 was prepared from 2.2 according to general method B in 57% yield. MS analysis m/z = 370+1.
Preparation ofN-(5-bromo-4-(morpholinomethyI)pyridin-2-yl)-2,2-dimethyl-butan-amide
(2A)

[0213] N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide 2A was prepared from 2.3 and morpholine bl following general method C in 36% yield.
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.67 (q, J= 8 Hz, 3H), 2.55 (m, 4H), 3.55 (s, 2H), 3.75 (m, 4H), 7.93 (s, 1H), 8.31 (s, 1H), 8.40 (s, 1H). MS analysis m/z = 370+1.
EXAMPLE 2B:
Preparation of 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyI)pyridin-2-yl)-butan-amide
(2B)
M Z Cl

[0214] 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide 2B was prepared according to general method D from 2A in 84% yield.
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.30 (s, 3H), 2.46 (m, 4H), 3.44 (s, 2H), 3.70 (m, 4H), 7.89 (s, 1H), 8.02 (s, 1H), 8.17 (s, 1H). MS analysis m/z = 305+1.
EXAMPLE 2C
Preparation of 2,2-dimethyl-N-(4-(morpholinomethyl)-3,3'-bipyridin-6-yl)butanamide (2C)
1C

[0215] Example 2C was prepared from 2 A following general method E in 20% yield.
1H NMR (CDCl3), δ 0.94 (t, J= 7 Hz, 3H), 1.31 (s, 6H), 1.70 (q, J= 7 Hz, 2H), 2.37 (br s, 4H), 3.37 (s, 2H), 3.62 (br s, 4H), 7.37 (q, J= 5 Hz and 2 Hz, 1H), 7.78 (d, J= 8 Hz, 1H), 8.03 (s, 1H), 8.15 (s, 1H), 8.38 (s, 1H), 8.64 (d, J= 5 Hz, 1H), 8.67 (s, 1H). MS analysis m/τ = 368+1.
[0216] Examples 2D - 2E were prepared from 2A using 4-pyridine boronic acid c3 and ethyl boronic acid c4 respectively following general method E.
2D: 1H NMR (CDCl3), δ 0.94 (t, J= 8 Hz, 3H), 1.31 (s, 6H), 1.70 (q, J= 7 Hz, 2H), 2.38 (t, J= 4 Hz, 4H), 3.39 (s, 2H), 3.62 (t, J= 5 Hz, 4H), 7.39 (br s, 1H), 8.03 (s, 1H), 8.15 (s, 1H), 8.39 (s, 1H), 8.68 (br s, 1H).
2E: 1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.22 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.46 (t, J= 5 Hz, 4H), 2.70 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.88 (s, 1H), 8.05 (s, 1H), 8.19 (s, 1H).
[0217] Examples 2F - 21 were prepared from the appropriate starting materials and following general methods as described in examples 2A and 2B.
2F: 1H NMR (CD3OD), δ 0.90 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.36 (m, 1H), 1.70 (q, J= 8 Hz, 2H), 1.85 (t, J= 5 Hz, 3H), 2.26 (t, J= 12 Hz, 2H), 2.82 (d, J= 2 Hz, 2H), 3.55 (s, 2H), 4.37 (s, 2H), 8.50 (s, 1H), 8.90 (s, 1H), 9.01 (s, 1H).
2G: 1H NMR (CD3OD), δ 0.93 (t, J= 8 Hz, 3H), 1.35 (s, 6H), 1.38 (t, J= 6 Hz, 1H), 1.57 (m, 1H), 1.81 (q, J= 8 Hz, 2H), 1.95 (m, 4H), 3.14 (m, 2H), 3.53 (d, J= 12 Hz, 2H), 4.58 (s, 2H), 7.86 (t, J= 3 Hz, 1H), 8.12 (s, 1H), 8.47 (d, J= 6 Hz, 1H).
2H: 1H NMR (CD3OD), δ 0.92 (m, 4H), 1.30 (m, 8H), 1.76 (d, J= 7 Hz, 2H), 3.00 (s, 3H), 3.31 (s, 1H), 3.59 (br s, 2H), 3.85 (br s, 2H), 4.59 (br s, 1H), 4.80 (br s, 1H), 8.37 (s, 1H), 8.65 (s, 1H).
21: 1H NMR (CDCl3), δ 1.32 (s, 9H), 2.29 (s, 3H), 2.46 (t, J= 5 Hz, 4H), 3.44 (s, 2H), 3.70 (t, 4H), 7.92 (br s, 1H), 8.02 (s, 1H), 8.17 (s, 1H).
EXAMPLE 23:
Preparation of N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide
(2J)

[0218] In a microwave vial, N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl- butanamide (2A, 0.5 g, 1.35 mmol), copper(I) bromide (0.387 g, 2.70 mmol), and sodium methoxide (c5, 1.890 g, 0.03498 mol, 25 weight% in MeOH) were added together. The vial was sealed, evacuated and heated in the microwave at 100 °C for 90 minutes. LC/MS indicated that the methoxy group was added, although the amide was cleaved off. The crude product was taken up in methanol and filtered through celite. The solvent was evaporated and the residue purified on a silica gel column using DCM/MeOH (0-20%) as eluent. The amine obtained was then dissolved in DCM (10 mL) and TEA was added (2.5 eq). Dimethyl butyric acid chloride a2 was then added (1.2 eq). The reaction was left to stir overnight. The reaction was quenched with ammonium chloride and the organic layer was separated off. The DCM layer was washed with brine, dried over magnesium sulfate, and the solvent evaporated. The crude product was purified on a silica gel column using ethyl acetate/hexane (10-70%) as eluent (yield 12%).
1H NMR (CDCl3), δ 0.92 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.55 (br s, 4H), 3.58 (br s, 2H), 3.77 (br s, 4H), 3.89 (s, 3H), 7.87 (br s, 2H), 8.27 (s, 1H). MS analysis m/z = 321+1.
EXAMPLE 2K:
Preparation of N-(5-(2-methoxyethylamino)-4-(morpholinomethyl)pyridin-2-yl)pivalamide
(2K)

1H NMR (DMSO), δ 1.23 (s, 9H), 3.16 (br s, 4H), 3.28 (s, 3H), 3.34 (t, J= 6 Hz, 2H), 3.56 (t, J = 6 Hz, 2H), 3.81 (br s, 4H), 4.30 (br s, 2H), 7.85 (s, 1H), 7.98 (s, 1H), 9.94 (br s, 1H).
[0220] Examples 2L - 2R were prepared from the appropriate starting materials and following general methods as described in examples 2A and 2B.
2L: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.26 (s, 3H), 3.03 (m, 4H), 3.10 (m, 4H), 3.61 (s, 2H), 7.91 (br s, 1H), 8.04 (s, 1H), 8.23 (s, 1H).
2M: 1H NMR (CDCl3), δ 1.77 (s, 3H), 3.11 (m, 4H), 3.16 (m, 4H), 3.75 (s, 2H), 8.34 (s, 1H), 8.38 (br s, 1H), 8.39 (s, 1H).
2N: 1H NMR (CDCl3), δ 1.77 (s, 3H), 2.28 (s, 3H), 3.04 (m, 4H), 3.13 (m, 4H), 3.63 (s, 2H), 8.11 (s, 1H), 8.16 (s, 1H), 8.32 (br s, 1H).
2O: 1H NMR (CDCl3), δ 0.50 (m, 2H), 0.64 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 2.27 (s, 3H), 3.03 (m, 4H), 3.10 (m, 4H), 3.61 (s, 2H), 8.06 (s, 1H), 8.23 (s, 1H), 8.50 (br s, 1H).
2P: 1H NMR (CDCl3), δ 0.92 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 2.73 (m, 4H), 2.80 (m, 4H), 3.56 (s, 2H), 7.94 (br s, 1H), 8.29 (s, 1H), 8.38 (s, 1H).
2Q: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.27 (s, 3H), 2.67 (m, 4H), 2.72 (m, 4H), 3.44 (s, 2H), 7.94 (br s, 1H), 8.00 (s, 1H), 8.17 (s, 1H).
2R: 1H NMR (CDCl3), δ 0.86 (t, J= 7 Hz, 3H), 1.23 (s, 6H), 1.64 (q, J= 7 Hz, 2H), 1.86 (m, 2H), 2.24 (s, 3H), 2.67 (m, 4H), 3.61 (s, 2H), 3.65 (t, J= 5 Hz, 2H), 3.75 (t, J= 6 Hz, 2H), 8.01 (s, 1H), 8.12 (br s, 1H), 8.15 (s, 1H).
EXAMPLE 2S:
Preparation of 6-(2, 2-dimethylbutanamido)-4-(morpholinomethyl)nicotinic acid (2.5)
2.5

[0221] To a solution of A/-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl- butanamide (2A, 3.71 g, 10.0 mmol) in DMF (30 mL) was added methanol (20 mL, 0.5 mol), triethylamine (2.23 mL, 0.0160 mol), 1,3-bis(diphenylphosphino)propane (dppp, 0.33 g, 0.80 mmol) and palladium acetate (0.18 g, 0.80 mol). Carbon monoxide was introduced and bubbled through the reaction mixture at -65 -70 °C for 4 hours. LC/MS showed the desired product peak and the starting bromide peak in ~ 1:1.3 ratio. The reaction mixture was then stirred at 65 °C - 70 °C for two additional hours, LC/MS showed the product : starting material ration ~ 1 : 1. More Pd(OAc)2 (90 mg, 0.04 eq.) and dppp (165 mg, 0.4 eq.) were added and the reaction mixture was stirred at 65 - 70 °C overnight with CO bubbling through the reaction solution. TLC showed the product : starting material ratio — 1.3:1. More Pd(OAc)2 (90 mg, 0.04 eq.), dppp (165 mg, 0.4 eq.) and TEA (2.23 mL, 1.6 eq.) were added and the reaction mixture was stirred at 65 - 70 °C for additional 6 hours. The reaction mixture was cooled to room temperature and diluted with EtO Ac-ether (1 :3), washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using EtOAc-hexane (1:1) as eluent, yielding the 1.90 g of the methyl ester.
[0222] To a solution of the methyl ester (1.80 g, 5.15mmol) in THF-MeOH-H2O (50-50-50 mL) was added LiOH monohydrate (1.68 g, 40 mmol). The reaction was stirred at room temperature overnight, and evaporated to remove solvent, acidified with 3N HCl to pH ~ 7 and extracted with DCM. The organic extracts were dried over sodium sulfate, concentrated in vacuo to give 1.7 g (51%) of the title acid 2.5. MS analysis m/z = 335-1.
Preparation ofN-(5-amino-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide
(2S)

1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.26 (s, 6H), 1.65 (q, J= 7 Hz, 2H), 2.44 (br s, 4H), 3.53 (br s, 2H), 3.69 (m, 4H), 4.65 (s, 2H), 7.69 (s, 1H), 7.76 (br s, 1H), 7.99 (s, 1H).
[0224] Examples 2T - 2V were prepared from the appropriate starting materials and following general methods as described in example 2A.
2T: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.67 (q, J= 7 Hz, 2H), 2.31 (s, 3H), 2.49 (br s, 4H), 2.59 (br s, 4H), 3.57 (s, 2H), 7.92 (br s, 1H), 8.29 (s, 1H), 8.37 (s, 1H).
2U: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.27 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.14 (s, 3H), 3.10 (t, J= 5 Hz, 2H), 3.17 (t, J= 4 Hz, 2H), 3.78 (t, J= 5 Hz, 2H), 3.91 (br s, 2H), 4.24 (s, 2H), 8.43 (s, 1H), 8.46 (br s, 1H), 8.59 (s, 1H).
2V: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.27 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.05 (d, J= 14 Hz, 2H), 2.70 (m, 2H), 2.95 (m, 2H), 3.81 (m, 2H), 4.45 (s, 2H), 7.18 (d, J= 7 Hz, 2H), 7.24 (t, J= 4 Hz, 1H), 7.32 (t, J= 7 Hz, 2H), 8.08 (br s, 1H), 8.48 (s, 1H), 8.60 (s, 1H).
EXAMPLE 3A:
Preparation of 5-methyl-4-(morpholinomethyl)pyridin-2-amine (3.1)

[0225] In a round bottom flask, 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2- yl)butanamide 2B (2.5 g, 8.186 mmol) was dissolved in 300 mL of 6.0 M HCl. The reaction was heated to reflux and left to stir overnight. The reaction was cooled to room temperature and then concentrated. The residue was basified with sodium bicarbonate and extracted with
ethyl acetate. The ethyl acetate layer was dried over magnesium sulfate and evaporated to yield the desired compound in 82% yield. MS analysis m/z = 207+1.
Preparation of 2-methyI-7V-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclohexanecarboxamide (3A)

[0226] Compound 3A was prepared from 3.1 and acid chloride al following general method A. 1H NMR (CDCl3), δ 0.94 (d, J= 6 Hz, 3H), 1.25 (m, 1H), 1.35 (m, 1H), 1.55 (m, 1H), 1.76 (m, 6H), 1.93 (m, 1H), 2.29 (s, 3H), 2.47 (t, J= 4 Hz, 4H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.88 (br s, 1H), 8.01 (s, 1H), 8.18 (s, 1H). MS analysis m/z = 331+1.
[0227] Example 3B was prepared from 3.1 and acid chloride a4 following general method A.
1H NMR (CD3OD), δ 1.31 (s, 6H), 1.36 (s, 6H), 1.49 (s, 1H), 2.52 (s, 3H), 3.34 (s, 2H), 3.48 (br s, 4H), 4.91 (br s, 4H), 7.93 (s, 1H), 8.23 (s, 1H). MS analysis m/z = 331+1.
[0228] Examples 3C - 3H were prepared from 3.1 and the appropriate acid chloride following general method A.
3C: 1H NMR (CDCl3), δ 0.83 (d, J= 7 Hz, 1H), 1.03 (d, J= 7 Hz, 2H), 1.22 (s, 1H), 1.44 (m, 2H), 1.56 (m, 2H), 1.68 (m, 2H), 1.78 (m, 2H), 2.00 (m, 1H), 2.30 (s, 3H), 2.46 (t, J= 4 Hz, 4H), 3.44 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.83 (br s, 1H), 8.01 (s, 1H), 8.14 (s, 0.7H), 8.19 (s, 0.3H).
3D: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.87 (m, 3H), 1.95 (m, 3H), 2.29 (s, 3H), 2.48 (m, 6H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.65 (br s, 1H), 8.01 (s, 1H), 8.20 (s, 1H). Anal. Calcd (C18H27N3O22HC1): C 54.55, H 7.54, N 10.60. Found; C 54.31, H 7.51, N 10.38
3E: 1H NMR (CDCl3), δ 0.87 (t, J= 8 Hz, 3H), 1.43 (m, 2H), 1.61 (m, 8H), 2.07 (m, 2H), 2.30 (s, 3H), 2.47 (t, J= 4 Hz, 4H), 3.44 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.92 (br s, 1H), 8.02 (s, 1H), 8.19 (s, 1H).
3F: 1H NMR (CDCl3), δ 1.06 (s, 6H), 1.20 (s, 6H), 1.36 (s, 3H), 2.29 (s, 3H), 2.47 (t, J= 5 Hz, 4H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.71 (br s, 1H), 8.00 (s, 1H), 8.14 (s, 1H).
3G: 1H NMR (CDCl3), δ 0.84 (t, J= 7 Hz, 9H), 1.67 (q, J= 8 Hz, 6H), 2.30 (s, 3H), 2.46 (t, J= 5 Hz, 4H), 3.44 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.92 (br s, 1H), 8.02 (s, 1H), 8.18 (s, 1H).
3H: 1H NMR (CDCl3), δ 0.72 (dd, J= 7 Hz and 4 Hz, 2H), 1.10 (t, J= 7 Hz, 3H), 1.25 (dd, J= 7 Hz and 4 Hz, 2H), 1.72 (q, J= 7 Hz, 2H), 2.29 (s, 3H), 2.46 (t, J= 4 Hz, 4H), 3.43 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 8.02 (br s, 2H), 8.14 (s, 1H).
Examples 31 - 3O were prepared from 3.1 and acids el- e9 utilizing the coupling reagent Bop-Cl (bis(2-oxo-3-oxazolidinyl)phosphinic chloride).
31: 1H NMR (CD3OD), δ 1.94 (s, 3H), 2.51 (s, 3H), 3.48 (m, 4H), 3.98 (m, 4H), 4.56 (s, 2H), 8.24 (s, 1H), 8.43 (s, 1H).
3 J: 1H NMR (CDCl3), δ 0.25 (m, 1H), 0.39 (m, 1H), 0.69 (m, 2H), 1.00 (m, 1H), 1.34 (d, J= 7 Hz, 3H), 1.67 (m, 1H), 2.29 (s, 3H), 2.47 (t, J= 5 Hz, 4H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 8.02 (s, 1H), 8.13 (br s, 1H), 8.20 (s, 1H).
3K: 1H NMR (CDCl3), δ 0.49 (m, 2H), 0.63 (m, 2H), 1.13 (s, 6H), 2.30 (s, 3H), 2.47 (t, J= 4 Hz, 4H), 3.44 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 8.03 (s, 1H), 8.18 (s, 1H), 8.47 (br s, 1H).
3L: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 6H), 1.23 (m, 2H), 1.55 (s, 3H), 1.75 (m, 2H), 2.30 (s, 3H), 2.46 (t, J= 5 Hz, 4H), 3.44 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.86 (br s, 1H), 8.02 (s, 1H), 8.17 (s, 1H).
3M: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.61 (m, 4H), 1.71 (m, 5H), 2.15 (m, 2H), 2.29 (s, 3H), 2.46 (t, J= 4 Hz, 4H), 3.44 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.82 (br s, 1H), 8.01 (s, 1H), 8.16 (s, 1H).
3N: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.32 (m, 2H), 1.58 (m, 2H), 2.30 (s, 3H), 2.46 (t, J= 4 Hz, 4H), 3.43 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.88 (br s, 1H), 8.02 (s, 1H), 8.17 (s, 1H).
3O: 1H NMR (CDCl3), δ 1.45 (s, 6H), 2.30 (s, 3H), 2.47 (t, J= 5 Hz, 4H), 2.58 (q, J= 11 Hz, 2H), 3.45 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 8.01 (br s, 1H), 8.03 (s, 1H), 8.14 (s, 1H).
EXAMPLE 4A:
Preparation of l-tert-butyl-3-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)urea (4A)

[0229] In a round bottom flask under nitrogen, 5-methyl-4-morpholin-4-ylmethylpyridin-2- ylamine (3.1, 200 mg, 1 mmol) was dissolved in DMF (10 mL, 0.1 mol). Sodium hydride (0.028 g, 1.2 mmol) was added and the solution was left to stir for 20 minutes at room temperature. The solution was then cooled to at 0 0C and left to stir for 1 hour. The solution became pink/red in color. ført-Butyl isocyanate (0.1653 mL, 1.447 mmol) was then added drop
wise to the solution. After aqueous work-up the crude product was purified on a silica gel column using ethylacetate/hexane (10-50%) eluents and 4A was obtained in 60 % yield.
1H NMR (CDCl3), δ 1.44 (s, 9H), 2.21 (s, 3H), 2.45 (t, J= 5 Hz, 4H), 3.38 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 6.72 (s, 1H), 7.30 (s, 1H), 7.92 (s, 1H), 9.30 (s, 1H). MS analysis m/x = 306+1.
EXAMPLE 5A;
Preparation of N-(5-bromo-4,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide (5.2)

[0230] N-(5-bromo-4,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide 5.2 was obtained from 5-bromo-4,6-dimethylpyridin-2-amine 5.1 according to general method A in 97% yield. MS analysis m/z = 298+1.
Preparation of7V-(5-bromo-4-(bromomethyl)-6-methylpyridin-2-yl)-2,2-dimethyl- butanamide (5.3) and N-(5-bromo-6-(bromomethyl)-4-methylpyridin-2-yl)-2,2- dimethylbutanamide (5.4)

[0231] N-(5-bromo-4-(bromomethyl)-6-methylpyridin-2-yl)-2,2-dimethyl-butanamide 5.3 and N-(5-bromo-6-(bromomethyl)-4-methylpyridin-2-yl)-2,2-dimethylbutanamide 5.4 were obtained as a mixture when N-(5-bromo-4,6-dimethylpyridin-2-yl)-2,2-dimethyl-butanamide 5.2 was reacted with NBS/AIBN following general procedure B. A separation at this stage was not possible.
Preparation of N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2- dimethylbutanamide (5A)
SB

[0232] The crude product mixture 5.3 and 5.4 (~0.9 mol) was dissolved in THF (50 mL), sodium bicarbonate (1.22 g, 14.6 mmol) was added followed by addition of morpholine (bl, 6.36 mL, 72.9 mol). The reaction mixture was stirred at room temperature for 3 days. The reaction mixture was diluted with EtOAc and washed with water. The organic layer was dried and concentrated. The residue was purified twice by column chromatography using acetone-hexane (1:80 - 1 :1), then EtOAc-heptane 1:3 as eluents to give pure compound 5A (210 mg, 6 % yield).
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.67 (q, J= 7 Hz, 2H), 2.55 (t, J= 5 Hz, 4H), 2.58 (s, 3H), 3.55 (s, 2H), 3.75 (t, J= 5 Hz, 4H), 7.87 (s, 1H), 8.20 (s, 1H) MS analysis m/z = 383+1.
[0233] Example 5B was also obtained from this reaction (2.0 g, 61% yield).
1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.42 (s, 3H), 2.60 (t, J= 4 Hz, 4H), 3.75 (m, 6H), 7.98 (s, 1H), 8.14 (s, 1H). MS analysis m/z = 383+1.
EXAMPLE 5C:
Preparation ofN-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl- butanamide (5C)

[0234] 7V-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide 5C was obtained from 5A according to general method D in 57% yield.
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.24 (s, 3H), 2.43 (s, 3H), 2.45 (t, J= 4 Hz, 4H), 3.42 (s, 2H), 3.69 (t, J= 5 Hz, 4H), 7.83 (s, 1H), 8.00 (s, 1H). MS analysis m/z = 319+1.
[0235] Example 5D, 2,2-dimethyl-N-(6-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide, was obtained as a minor product (28% yield) from the same reaction.
1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.44 (m, 7H), 3.45 (s, 2H), 3.72 (t, J= 5 Hz, 4H), 6.94 (s, 1H), 7.92 (s, 1H), 8.03 (s, 1H). MS analysis m/z = 305+1.
EXAMPLE 5E;
Preparation of N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl- butanamide (5E)

[0236] N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide 5E was obtained from 5B according to general method D in 85% yield.
1H NMR (CDCl3), δ 0.90 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.25 (s, 3H), 2.29 (s, 3H), 2.48 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.87 (s, 1H), 8.02 (s, 1H). MS analysis m/z = 319+1.
EXAMPLE 6A;
Preparation of 7V-(5-bromo-6-methylpyridin-2-yl)-2,2-dimethylbutanamide (6.2)

[0237] N-(5-bromo-6-methylpyridin-2-yl)-2,2-dimethylbutanamide 6.2 was prepared from commercially available 5-bromo-6-methylpyridin-2-amine 6.1 and 2,2-dimethyl-butanoyl chloride al in 99% yield following general method A. MS analysis m/z = 285+1.
Preparation ofN-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide
(6A)

1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.60 (t, 4H), 3.71 (s, 2H), 3.75 (t, J= 5 Hz, 4H), 7.81 (d, J= 9 Hz, 1H), 8.04 (s, 1H), 8.09 (d, J= 9 Hz, 1H). MS analysis m/z = 370+1.
[0239] Example 6B was obtained from 6A in 76% yield following general method D.
1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.34 (s, 3H), 2.49 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.45 (d, J= 8 Hz, 1H), 7.93 (s, 1H), 8.06 (d, J= 8 Hz, 1H). MS analysis m/z = 305+1.
EXAMPLE 7A:
Preparation of N-(4,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide (7.2)

[0240] N-(4,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide 7.2 was prepared from 4,6- dimethylpyridin-2-amine 7.1 and 2,2-dimethylbutanoyl chloride al following general method A in 95 % yield. MS analysis m/z = 220+1.
Preparation of 2,2-dimethyl-N-(4-methy--6-(morpholinomethyl)pyridin-2-yl)butan-amide
(7A)

[0241] 2,2-dimethyl-N-(4-methyl-6-(morpholinomethyl)pyridin-2-yl)butanamide 7A was prepared from 7.2 according to general method B, followed by general method C in 46% overall yield.
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.34 (s, 3H), 2.49 (t, J= 4 Hz, 4H), 3.50 (s, 2H), 3.75 (t, 4H), 6.94 (s, 1H), 7.94 (s, 1H), 8.03 (s, 1H). MS analysis m/z = 305+1.
EXAMPLE 8A:
Preparation of 2,4-dichloroquinoline (8.2)

[0242] A suspension of the 4-hydroxyquinolin-2(1H)-one (8.1, 5g, 31 mmol) in POCl3 (28.9 mL, 310 mmol)was heated at 85 °C for 2.5 hours. Following a procedure described in
EP 1464335 A2, the reaction was cooled and ice was carefully added until a precipitate formed.
The mixture was stirred for 1 hour, the precipitate (starting material) was filtered off and DCM was added. The layers were separated and the organic phase was concentrated and purified by a silica gel plug, rinsing with 10-20% ethyl acetate/hexanes, yielding 3.95 g of 8.2 as a light orange solid (64%).
MS analysis m/z = 198+1.
Preparation of 2-chloro-4-morpholin-4-yl-quinoline (8.3)

[0243] To a solution of the 8.2 (2.0g, 10.1 mmol) and TEA (4.22mL, 30.3 mmol) in DMF (20mL) under nitrogen was added morpholine (bl, 1.76mL, 20.2 mmol). The reaction was stirred at 60 °C for 3 days. The mixture was concentrated, dissolved in DCM and washed with water. The organic layer was concentrated and purified by a flash silica gel column using an ethyl acetate/hexanes (5-30%) gradient to give 723 mg (29%) 8.3 as an off-white solid. MS analysis m/z = 248+1.
Preparation of 4-morpholinoquinolin-2-amine (8.4)

[0244] A mixture of 8.3 (0.4 g, 1.61 mmol) and aqueous ammonium hydroxide (5mL, 128 mmol) was heated at 160 °C in a sealed vessel for 20 hours. The reaction was cooled and water and DCM were added. The layers were separated and the organic layer was concentrated and purified by a silica gel plug using a 2-10% methanol/DCM (plus NH4OH) gradient. The solid obtained was triturated in ethyl acetate, filtered and dried to give 90 mg of 8.4 (24% yield) as an orange solid. MS analysis m/z = 230+1.
Preparation of 2,2-dimethyl-N-(4-morpholinoquinolin-2-yl)butanamide dihydro-chloride salt (8A)

[0245] To a solution of 8.4 (30 mg, 0.126 mmol) in DCM (5mL) under nitrogen was added TEA 88 ul, 0.633mmol), then the acid chloride a2 (35 ul, 0.253mmol). The reaction was stirred at room temperature for 5 hours. The reaction was diluted with water and layers were separated. Organics were concentrated and purified by a silica gel plug using a 20-30% ethyl acetate/hexanes gradient. The product was stirred with 2 mL of IM HCl/Et2O and concentrated. The solid obtained was triturated in diethyl ether (with 2 drops DCM), filtered and dried to give 15 mg light yellow solid 8A (29%).
1H NMR (CD3OD), δ 0.95 (t, J= 7 Hz, 3H), 1.35 (s, 6H), 1.80 (q, J= 7 Hz, 2H), 3.69 (br s, 4H), 3.98 (br s, 4H), 7.12 (s, 1H), 7.70 (m, J= 9 Hz, 1H), 7.91 (t, J= 7 Hz, 1H), 8.00 (d, J= 8 Hz, 1H), 8.14 (d, J= 8 Hz, 1H). MS analysis m/z = 328+1.
[0246] Example 8B was obtained from 8.4 and 2-methylcyclohexanecarbonyl chloride al following general method A in 76% yield.
1H NMR (CDCl3), δ 1.01 (d, J= 7 Hz, 3H), 1.59 (m, 3H), 1.72 (m, 2H), 1.84 (m, 3H), 2.28 (m, 1H), 2.52 (m, 1H), 3.29 (t, J= 5 Hz, 4H), 3.98 (t, J= 5 Hz, 4H), 7.38 (m, 1H), 7.60 (m, 1H), 7.75 (dd, J= 7 Hz and 1 Hz, 1H), 7.94 (dd, J= 8 Hz and 1 Hz, 1H), 8.04 (s, 1H).
EXAMPLE 9A:
Preparation of 4-(bromomethyl)-2-chloroquinoline (9.2)

[0247] 4-(Bromomethyl)-2-chloroquinoline 9.2 was prepared according to a procedure by Otsubo (Chemical & Pharmaceutical Bulletin 1991, 39(11), 2906-9).
Preparation of 4-((2-chloroquinolin-4-yl)methyl)morpholine (9.3)

[0248] Compound 9.2 (2g, 7.8 mmol) was dissolved in DCM (60 mL) under nitrogen and TEA (2.72mL, 19.5 mmol) and morpholine bl (1.36 mL, 15.6 mmol) were added. The reaction was stirred at room temperature for 2 days. Water was added and the layers were separated. The organic layer was concentrated and triturated in ethyl acetate. The filtrate was concentrated and triturated in diethyl ether/hexane (9:1). Solids were filtered to give 1.10 g light orange solid 9.3. The filtrate was concentrated and triturated in hexanes, filtered and dried to give additional 0.61 g light orange solid 9.3 (total yield = 83%). MS analysis m/z = 262+1.
Preparation of 4-(morpholinomethyl)quinolin-2-amine (9.4)

Preparation of 2,2-dimethyl-7V-(4-(morpholinomethyl)quinolin-2-yl)butanamide (9)

[0250] To a solution of the amine 9.4 (30 mg, 0.123 mmol) in DCM (5mL) under nitrogen was added TEA (51.6 uL, 0.136 mmol), then the acid chloride a2 (18.6 uL, 0.136 mmol) . The reaction was stirred at room temperature for 16 hours. The reaction was concentrated, dissolved in ethyl acetate and purified by a silica gel plug, rinsing with 20-40% ethyl acetate/hexanes. The fractions were concentrated and dried to give 40 mg 9 A as a light orange solid (92% yield).
1H NMR (CDCl3), d 0.95 (t, J= 7 Hz, 3H), 1.34 (s, 6H), 1.72 (q, J= 7 Hz, 2H), 2.54 (t, J= 4 Hz, 4H), 3.71 (t, J= 5 Hz, 4H), 3.90 (s, 2H), 7.46 (m, 1H), 7.66 (m, 1H), 7.82 (d, J= 8 Hz, 1H), 8.17 (s, 1H), 8.25 (t, J= 9 Hz, 1H), 8.46 (s, 1H). MS analysis m/z = 341+1.
EXAMPLE 10A;
Preparation of 2-chloro-6,7-dihydro-5H-cyclopenta[b]pyridine-4-carbonyl chloride (10.4a)

Preparation of (I-chloro-β^-dihydro-SH-cyclopentalb]pyridin^-yl)(morpholino)- methanone (10.5a)

[0252] Morpholine bl (20 mL, 0.2 mol) was added to 2-chloro-6,7-dihydro-5H-[b]pyrindine-4- carbonyl chloride 10.4a (6 g, 0.03 mol) in toluene (100 mL) at 0°C. The mixture was allowed to warm up to RT and stirred overnight. The reaction was diluted with ethyl acetate (100ml), washed with water (100ml), dried over MgSO4 and concentrated under vacuum. The crude product was purified by flash chromatography on SiO2 using 0-60% ethyl acetate in hexane to give the product 10.5a (2.22g, 30% yield) as a yellow solid. MS analysis m/z = 266+1.
Preparation of 4-((2-chloro-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)methyl)-morpholine (10.6a)

[0253] Borane-dimethyl sulfide complex (8.2 mL, 2M in THF) was added to (2-chloro-6,7- dihydro-5H-[b]pyrindin-4-yl)-morpholin-4-yl-methanone (10.5a, 2.2 g, 0.0082 mol) in THF (50 mL). The reaction was heated to reflux for 3h, then cooled to RT. MeOH (20ml) was added slowly and the mixture was stirred for lhr. Hydrochloric acid (20ml, 4M in dioxane) was added and the reaction was heated to reflux for lhr, then concentrated under vacuum. The residue was diluted with sat. sodium bicarbonate (50ml) and extracted with ethyl acetate (2X50ml). The organic layers were combined, dried over MgSO4 and concentrated under vacuum. The crude product was purified by flash chromatography on SiO2 using 0-60% ethyl acetate in heptanes to give the product 10.6a as colorless oil (1.62g, 77% yield). MS analysis m/z - 252+1.
Preparation of 4-(morpholinomethyl)-6J-dihydro-5H-cyclopenta[b]pyridin-2-amine (10.7a)

[0254] 4-((2-Chloro-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)methyl)-morpholine 10.6a (0.50 g, 0.0020 mol) was added to hydrazine (5 mL, 0.2 mol) and heated to reflux overnight. The mixture was concentrated under vacuum to give a white solid. This crude intermediate and a Raney Nickel slurry in water (0.1 g , nickel : water 1:1) were added to methanol (20 mL, 0.5 mol) under nitrogen and placed in Parr shaker at 10 psi of hydrogen for lhr. The mixture was filtered over celite and the celite washed with methanol (20ml). The filtrates were concentrated under vacuum. The residue was dissolved in methylene chloride (30ml), washed with brine (20ml) and concentrated under vacuum to give a white solid. The crude product 10.7a (180mg, 39% yield) was used in the next step. MS analysis m/z = 233+1.
Preparation of 2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta- [b]pyridin-2-yl)butanamide (10A)

[0255] 2,2-Dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide 10A was prepared from 10.7a and a2 in 60% yield according to general method A.
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.12 (t, J= 8 Hz, 3H), 2.45 (t, J= 5 Hz, 4H), 2.92 (t, J= 8 Hz, 4H), 3.43 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.88 (s, 1H), 8.04 (s, 1H). MS analysis m/z = 331+1.
[0256] Example 10B was prepared following the procedures described in example 10A from cyclohexanone d2.
10B: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.27 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 1.83 (m, 4H), 2.46 (t, J= 4 Hz, 3H), 2.75 (t, J= 6 Hz, 2H), 2.79 (t, J= 6 Hz, 2H), 3.38 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.92 (s, 1H), 8.02 (s, 1H).
[0257] Example 10C was prepared in a similar fashion to 10A using 2-butanone d3 and acid chloride al .
10C: 1H NMR (CDCl3), δ 0.98 (d, J= 7 Hz, 3H), 1.26 (m, 4H), 1.40 (m, 1H), 1.51 (m, 2H), 1.67 (m, 2H), 1.80 (m, 2H), 2.18 (m, 1H), 2.46 (t, J= 4 Hz, 4H), 2.69 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.73 (t, J= 5 Hz, 4H), 6.94 (s, 1H), 8.03 (s, 1H), 8.28 (s, 1H).
[0258] Examples 10D - 10L were prepared in a similar fashion to 10A using 10.7dl or 10.7dx and the appropriate acid chloride.
10D: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.86 (m, 3H), 1.94 (m, 3H), 2.12 (m, 2H), 2.47 (m, 6H), 2.91 (t, J= 8 Hz, 4H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.64 (br s, 1H), 8.06 (s, 1H).
10E: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.59 (m, 2H), 1.71 (m, 6H), 2.15 (m, 4H), 2.45 (t, J= 4 Hz, 4H), 2.92 (t, J= 8 Hz, 4H), 3.43 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.90 (br s, 1H), 8.03 (s, 1H).
10F: 1H NMR (CDCl3), δ 0.86 (t, J= 8 Hz, 3H), 1.42 (m, 5H), 1.61 (m, 5H), 2.12 (m, 4H), 2.46 (t, J= 4 Hz, 4H), 2.92 (t, J= 8 Hz, 4H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.95 (br s, 1H), 8.07 (s, 1H).
10G: 1H NMR (CDCl3), δ 0.84 (t, J= 7 Hz, 9H), 1.67 (q, J= 7 Hz, 6H), 2.11 (m, 2H), 2.45 (t, J = 4 Hz, 4H), 2.92 (t, J= 8 Hz, 4H), 3.43 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.91 (br s, 1H), 8.06 (s, 1H).
10H: 1H NMR (CDCl3), δ 1.00 (s, 1H), 1.16 (s, 6H), 1.31 (s, 6H), 2.11 (m, 2H), 2.43 (t, J= 4 Hz, 4H), 2.91 (m, 4H), 3.42 (s, 2H), 3.69 (t, J= 5 Hz, 4H), 7.89 (s, 1H), 8.34 (br s, 1H).
101: 1H NMR (CDCl3), δ 0.48 (m, 2H), 0.61 (m, 2H), 1.07 (m, 1H), 1.13 (s, 6H), 2.12 (m, 2H), 2.46 (t, J= 4 Hz, 4H), 2.92 (m, 4H), 3.44 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 8.04 (s, 1H), 8.42 (br s, 1H).
10J: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 1.91 (m, 2H), 2.12 (m, 2H), 2.69 (m, 4H), 2.92 (t, J= 8 Hz, 4H), 3.58 (s, 2H), 3.71 (m, 2H), 3.82 (t, J= 6 Hz, 2H), 7.90 (br s, 1H), 8.06 (s, 1H).
10K: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.59 (m, 2H), 1.70 (m, 6H), 1.91 (m, 2H), 2.11 (m, 2H), 2.18 (m, 2H), 2.69 (m, 4H), 2.92 (t, J= 8 Hz, 4H), 3.58 (s, 2H), 3.71 (t, J= 5 Hz, 2H), 3.82 (t, 2H), 7.92 (br s, 1H), 8.06 (s, 1H).
10L: 1H NMR (CDCl3), δ 0.84 (t, J= 7 Hz, 9H), 1.67 (q, J= 7 Hz, 6H), 1.91 (m, 2H), 2.12 (m, 2H), 2.69 (m, 4H), 2.92 (t, J= 8 Hz, 4H), 3.58 (s, 2H), 3.71 (m, 2H), 3.82 (t, J= 6 Hz, 2H), 7.95 (br s, 1H), 8.08 (s, 1H).
EXAMPLE IQM;
Preparation of7V-cycloheptyI-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta-
(b]pyridin-2-amine (10M)

[0259] To 2-chloro-4-morpholin-4-ylmethyl-6,7-dihydro-5H-[ 1]pyridine (10.θdl, 0.150 g, 0.593 mmol) was added cycloheptylamine (0.85 mL, 6.6 mmol). The mixture was heated to 200°C for 4hrs in the microwave. The sample was concentrated under vacuum and purified via flash chromatography using 50-100% ethyl acetate in hexane to yield 10M as a white solid (yield 13%).
1H NMR (CDCl3), δ 1.50 (m, 5H), 1.60 (m, 5H), 1.98 (m, 2H), 2.04 (m, 2H), 2.45 (t, J= 4 Hz, 4H), 2.77 (t, J= 7 Hz, 2H), 2.82 (t, J= 8 Hz, 2H), 3.35 (s, 2H), 3.62 (m, 1H), 3.72 (t, J= 5 Hz, 4H), 4.41 (d, J= 8 Hz, 1H), 6.16 (s, 1H)
[0260] Examples 10N - 10O were prepared in a similar fashion to 10M using 10.6dl and 1- methoxypropan-2-amine or (1R,2R,4S)-bicyclo[2.2.1]heptan-2-amine respectively.
10N: 1H NMR (CDCl3), δ 1.23 (d, J= 7 Hz, 3H), 2.05 (m, 2H), 2.44 (t, J= 4 Hz, 4H), 2.77 (t, J = 7 Hz, 2H), 2.83 (t, J= 8 Hz, 2H), 3.33 (s, 2H), 3.37 (s, 3H), 3.40 (m, 1H), 3.44 (m, 1H), 3.71 (t, J= 5 Hz, 4H), 3.96 (m, 1H), 4.49 (d, J= 8 Hz, 1H), 6.25 (s, 1H).
10O: 1H NMR (CDCl3), δ 1.21 (m, 4H), 1.51 (m, 3H), 1.83 (m, 1H), 2.04 (m, 2H), 2.25 (m, 2H), 2.46 (t, J= 4 Hz, 4H), 2.80 (m, 4H), 3.36 (s, 3H), 3.72 (t, J= 5 Hz, 4H), 4.40 (d, J= 7 Hz, 1H), 6.19 (s, 1H).
EXAMPLE 11A:
Preparation of N-((2-methyIcycIohexyI)methyl)-4-(morpholinomethyl)pyridin-2-amine
(11A)
 11A
11A
[0261] A 50 mL flask was charged with 2-methyl-N-(4-(morpholinomethyl)pyridinyl-2- yl)cyclohexanecarboxamide IA (140 mg, 0.44 mmol) and THF (2 mL). The solution was cooled in an ice water bath and the 1.0 M of borane in THF (3 mL) was added dropwise. After 30 minutes the cooling bath was removed and the reaction was allowed to stir for two hours at room temperature, then the reaction was heated at 40 0C overnight. The reaction was cooled in an ice/water bath and quenched with 2 mL of methanol added dropwise. After a 30 minutes 2 mL of 1.0 N HCL were added and after 15 minutes 3 mL of 6.0 N HCL were added. The reaction was allowed to stir overnight at 40 °C. After cooling in an ice water bath the reaction was made strongly basic by addition of 8 mL of 1.0 N NaOH and 400 mg of solid NaOH. Methanol and THF were removed under vaccum and the remaining aqueous phase was extracted with two 50 mL portions of ethyl acetate. The combined extracts were dried with sodium sulfate and concentrated under vacuum. The material was chromatographed on a silica column with an ethyl acetate/hexanes (25 to 100% gradient) yielding 27mg 11A (20% yield).
1H NMR (CDCl3), δ 0.97 (d, J= 7 Hz, 3H), 1.37 (m, 4H), 1.57 (m, 3H), 1.72 (m, 1H), 1.94 (m, 1H), 2.03 (m, 1H), 3.26 (m, 1H), 3.58 (m, 1H), 3.66 (m, 1H), 3.73 (m, 1H), 3.98 (br s, 4H), 4.41 (s, 2H), 7.06 (dd, J= 7 Hz and 2 Hz, 1H), 7.38 (m, 1H), 7.91 (t, J= 7 Hz, 1H). MS analysis m/z = 303+1.
EX MPLE HB:
Preparation ofiV-(2,2-dimethylbutyl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclo- penta[b]pyridin-2-amine (HB)
HB

[0262] 2,2-Dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)butanamide 1OA (0.15 g, 0.45 mmol) was dissolved in THF (5 mL). Borane-dimethyl sulfide
complex (0.4 mL, 2M in THF) was added and the mixture heated to reflux for 3h. The reaction was cooled to RT and methanol (2ml) was added slowly. The mixture was stirred for Ih, then 4M HCl in dioxane (3ml) was added and heated to reflux for lhr. The reaction was concentrated under vacuum, diluted with sat. sodium bicarbonate (20ml) and extracted with DCM (2X20ml). The organic layers were dried over MgSO4, concentrated under vacuum and purified by flash chromatography on SiO2 using 0-50% ethyl acetate in hexane to give the product HB as colorless oil (19 mg, 13% yield).
1H NMR (CDCl3), δ 0.85 (t, J= 8 Hz, 3H), 0.94 (s, 6H), 1.35 (q, J= 8 Hz, 2H), 2.06 (m, 2H), 2.45 (t, J= 4 Hz, 4H), 2.78 (t, J= 7 Hz, 2H), 2.84 (t, J= 8 Hz, 2H), 3.01 (d, J= 6 Hz, 2H), 3.34 (s, 2H), 3.72 (t, J= 5 Hz, 4H), 6.23 (s, 1H). MS analysis m/z = 317+1.
[0263] Examples 11C and 11D were prepared from 2B and 10C following the procedure described for example 11B.
HC: 1H NMR (CDCl3), δ 0.86 (t, J= 8 Hz, 3H), 0.93 (s, 6H), 1.34 (q, J= 8 Hz, 2H), 2.15 (s, 3H), 2.46 (t, J= 5 Hz, 4H), 3.06 (d, J= 6 Hz, 2H), 3.34 (s, 2H), 3.72 (t, J= 5 Hz, 4H), 6.42 (s, 1H), 7.81 (s, 1H).
HD: 1H NMR (CDCl3), δ 0.86 (t, J= 8 Hz, 3H), 0.94 (s, 6H), 1.24 (t, J= 8 Hz, 3H), 1.35 (q, J= 8 Hz, 2H), 2.45 (t, J= 4 Hz, 4H), 2.60 (q, J= 8 Hz, 2H), 3.01 (d, J= 6 Hz, 2H), 3.37 (s, 2H), 3.73 (t, J= 5 Hz, 4H), 6.21 (s, 1H), 6.42 (s, 1H).
EXAMPLE 14A:
Preparation of 2,2-dimethyI-Λ^4-(morpholinomethyl)-5-(prop-1-en-2-yl)pyridin-2- yl)butanamide (14.1. a)

[0264] To a solution of bromide 2 A (400mg, 1.08 mmol) in water/dioxane (2mL/6mL) was added potassium carbonate (448mg, 3.24 mmol), 4,4,5, 5-tetramethyl-2-(prop-1-en-2-yl)-1, 3,2- dioxaborolane (c6, 200mg, 1.19 mmol) and catalyst Pd(PPh3)4 (120 mg, 0.11 mmol). The reaction was microwaved at 120 °C for 15 minutes. The reaction was run through a plug of Celite, rinsing with ethyl acetate. The filtrate was concentrated down to give the crude product. The crude product was purified on a silica gel column using ethyl acetate/hexane (0-75%) as eluent (yield 97%). MS analysis m/z = 331+1.
Preparation of N-(5-isopropyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide (14A)
14A

[0265] In a round bottom flask, 2,2-dimethyl-N-(4-(morpholinomethyl)-5-(prop-1-en-2- yl)pyridin-2-yl)butanamide (14.1a, 210 mg, 0.6336 mmol) was dissolved in methanol (8 mL). Palladium on carbon (7.05 mg, 0.0576 mmol) was added and the flask was evacuated. Hydrogen (0.3 L, 0.01 mol) was added next with a balloon. The reaction was left to stir for 1 hour. The reaction was filtered through celite and the methanol was evaporated off to give the desired product (yield 67%).
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.27 (d, J= 7 Hz, 6H), 1.28 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 2.46 (m, 4H), 3.30 (m, 1H), 3.49 (s, 2H), 3.69 (t, J= 5 Hz, 4H), 7.96 (br s, 1H), 8.16 (s, 1H), 8.16 (s, 1H).
[0266] Example 14B was prepared from 2A and c7 following the procedure described for example 14 A.
1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.22 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 2.46 (t, J= 5 Hz, 4H), 2.70 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.88 (br s, 1H), 8.05 (s, 1H), 8.19 (s, 1H)
EXAMPLE 14C:
Preparation of N-(5-ethyl-4-(niorpholinomethyI)pyridin-2-yl)-2,2,3,3-tetramethyl- cyclopropanecarboxamide (14C)

[0267] Example 14 was prepared from 14B and a4 following e procedures described for example 3A.
1H NMR (CDCl3), δ 1.00 (s, 1H), 1.15 (m, 9H), 1.25 (s, 6H), 2.38 (t, J= 4 Hz, 4H), 2.63 (q, J- 7 Hz, 2H), 3.40 (s, 2H), 3.63 (t, J= 5 Hz, 4H), 7.95 (s, 1H), 8.04 (s, 1H), 8.21 (br s, 1H)
[0268] Example 14D - 14K were prepared from 2A and the appropriate acid chlorides following the procedures described for example 3A.
14D: 1H NMR (CDCl3), δ 1.06 (s, 6H), 1.20 (s, 6H), 1.22 (t, J= 7 Hz, 3H), 1.36 (s, 3H), 2.48 (t, J= 4 Hz, 4H), 2.69 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.63 (br s, 1H), 8.04 (s, 1H), 8.16 (s, 1H).
14E: 1H NMR (CDCl3), δ 0.49 (m, 2H), 0.63 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 1.22 (t, J= 8 Hz, 3H), 2.46 (t, J= 5 Hz, 4H), 2.70 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 8.06 (s, 1H), 8.20 (s, 1H), 8.47 (br s, 1H).
14F: 1H NMR (CDCl3), δ 0.90 (t, J= 8 Hz, 6H), 1.23 (s, 3H), 1.54 (m, 2H), 1.75 (m, 2H), 2.46 (t, J= 4 Hz, 4H), 2.70 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.88 (br s, 1H), 8.05 (s, 1H), 8.19 (s, 1H).
14G: 1H NMR (CDCl3), δ 0.84 (t, J= 7 Hz, 9H), 1.23 (t, J= 8 Hz, 3H), 1.67 (q, J= 7 Hz, 6H), 2.46 (m, 4H), 2.70 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.93 (br s, 1H), 8.05 (s, 1H), 8.20 (s, 1H).
14H: 1H NMR (CDCl3), δ 1.23 (t, J= 8 Hz, 3H), 1.77 (s, 3H), 2.48 (br s, 4H), 2.71 (q, J= 7 Hz, 2H), 3.49 (s, 2H), 3.72 (br s, 4H), 8.11 (s, 1H), 8.13 (s, 1H), 8.31 (br s, 1H).
141: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.22 (t, J= 8 Hz, 3H), 1.87 (m, 2H), 1.95 (m, 2H), 2.48 (m, 6H), 2.69 (q, J= 8 Hz, 2H), 3.48 (br s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.66 (br s, 1H),
8.04 (s, 1H), 8.22 (s, 1H).
14J: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.22 (t, J= 8 Hz, 3H), 1.61 (m, 2H), 1.70 (m, 6H), 2.18 (m, 2H), 2.46 (t, J= 4 Hz, 4H), 2.70 (q, J= 8 Hz, 2H), 3.47 (s, 2H), 3.70 (t, J= 5 Hz, 4H), 7.84 (br s, 1H), 8.04 (s, 1H), 8.18 (s, 1H).
14K: 1H NMR (CDCl3), δ 0.87 (t, J= 8 Hz, 3H), 1.23 (t, J= 7 Hz, 3H), 1.40 (m, 5H), 1.62 (m, 5H), 2.07 (m, 2H), 2.48 (br s, 4H), 2.71 (q, J= 8 Hz, 2H), 3.48 (s, 2H), 3.71 (t, J= 5 Hz, 4H),
8.05 (br s, 2H), 8.23 (s, 1H).
EXAMPLE 14L:
Preparation of 5-methoxy-4-(morpholinomethyl)pyridin-2-amine (14.2a)
2A 14.2a
 [0269] A mixture of N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide (2 A, 7.41 g, 0.0200 mol), copper© bromide (4.66 g, 0.0325 mol) and NaOMe (60 mL, 25 wt. % in MeOH) was heated at ~ 105 °C under nitrogen atmosphere for 24 hours. After cooling to room temperature, the mixture was quenched with diluted HCl to pH ~ 9-10 and extracted with EtOAc. The organic extracts were combined, washed with sat. NH4Cl, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using MeOH-DCM (1 :10) as eluent, yielding 3.0 g of the product 14.2a (yield 67%). MS analysis m/z = 223+1.
[0269] A mixture of N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide (2 A, 7.41 g, 0.0200 mol), copper© bromide (4.66 g, 0.0325 mol) and NaOMe (60 mL, 25 wt. % in MeOH) was heated at ~ 105 °C under nitrogen atmosphere for 24 hours. After cooling to room temperature, the mixture was quenched with diluted HCl to pH ~ 9-10 and extracted with EtOAc. The organic extracts were combined, washed with sat. NH4Cl, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using MeOH-DCM (1 :10) as eluent, yielding 3.0 g of the product 14.2a (yield 67%). MS analysis m/z = 223+1.
Preparation of N-(5-methoxy-4-(morpholinomethyI)pyridin-2-yl)-2,2,3,3-tetra- methylcyclopropanecarboxamide (14L)

[0270] 2,2,3,3-Tetramethylcylopropanecarboxylic acid (a4, 372 mg, 2.62 mmol), 5-methoxy-4- (morpholinomethyl)pyridin-2-amine (14.2a, 450 mg, 2.0 mmmol) and bis(2-oxo-3- oxazolidinyl)phosphinic chloride (BOP-Cl, 698 mg, 2.74 mmol) were dissolved in DCM (12 mL). To this reaction mixture was added Λ^V-diisopropylethylamine (0.878 mL, 5.04 mmol) dropwise under nitrogen atmosphere. The reaction was stirred at room temperature overnight. The reaction mixture was washed with sat. NaHCO3, dried over sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography using acetone-hexane-Et3N (10:40:5) as eluent, yielding 80 mg of the product, which was further purified by column chromatography using acetone-hexane (1:1) as eluent, yielding 76 mg (11%) of compound 14L.
1H NMR (CDCl3), δ 1.01 (s, 1H), 1.21 (s, 6H), 1.32 (s, 6H), 2.50 (t, J= 5 Hz, 4H), 3.52 (s, 2H), 3.72 (t, J= 5 Hz, 4H), 3.88 (s, 3H), 7.81 (br s, 1H), 7.84 (s, 1H), 8.14 (s, 1H)
[0271] Examples 14M - 14S were prepared from 14.2a and the appropriate acids following the procedure described for example 14L.
14M: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 6H), 1.23 (s, 3H), 1.54 (m, 2H), 1.76 (m, 2H), 2.52 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 3.88 (s, 3H), 7.85 (s, 1H), 7.86 (br s, 1H), 8.26 (s, 1H).
14N: 1H NMR (CDCl3), δ 0.85 (t, J= 8 Hz, 9H), 1.67 (q, J= 7 Hz, 6H), 2.52 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 3.88 (s, 3H), 7.85 (s, 1H), 7.87 (br s, 1H), 8.26 (s, 1H).
14O: 1H NMR (CDCl3), δ 1.76 (s, 3H), 2.52 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.76 (t, J= 5 Hz, 4H), 3.91 (s, 3H), 7.90 (s, 1H), 8.20 (s, 1H), 8.25 (br s, 1H).
14P: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.86 (m, 3H), 1.95 (m, 3H), 2.47 (m, 2H), 2.53 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.75 (t, J= 5 Hz, 4H), 3.89 (s, 3H), 7.62 (br s, 1H), 7.84 (s, 1H), 8.29 (s, 1H).
14Q: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.62 (m, 2H), 1.71 (m, 6H), 2.17 (m, 2H), 2.52 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 3.88 (s, 3H), 7.84 (br s, 2H), 8.26 (s, 1H).
14R: 1H NMR (CDCl3), δ 0.87 (t, J= 8 Hz, 3H), 1.41 (m, 5H), 1.60 (m, 5H), 2.07 (m, 2H), 2.52 (t, J= 5 Hz, 4H), 3.54 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 3.88 (s, 3H), 7.85 (s, 1H), 7.92 (br s, 1H), 8.28 (s, 1H).
14S: 1H NMR (CDCl3), δ 0.92 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 3.08 (m, 8H), 3.71 (s, 2H), 3.89 (s, 3H), 7.87 (s, 1H), 7.88 (br s, 1H), 8.28 (s, 1H).
EXAMPLE 14T:
Preparation of 5-ethoxy-4-(morphoIinomethyl)pyridin-2-amine (14.2b)
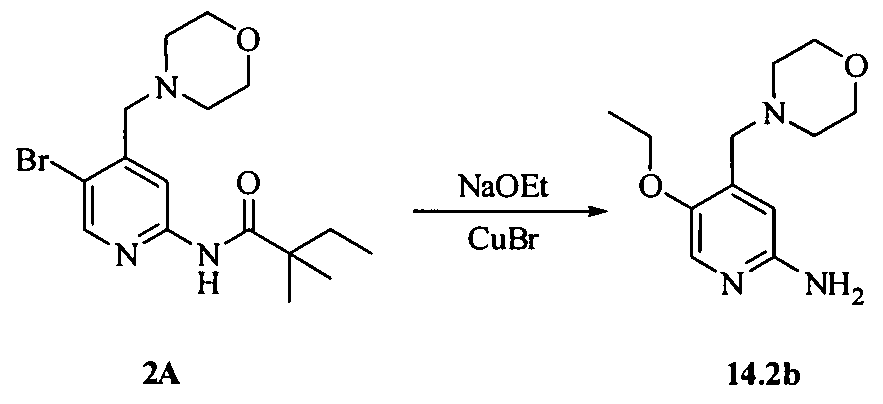
[0272] Into a flask charged with N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2- dimethylbutanamide (2A, 4.30 g, 11.6 mmol), copper(I) bromide (2.7 g, 19 mmol) and sodium ethoxide (120 mL, 0.32 mol). The reaction was refluxed at 105 0C for 24 hours under the nitrogen atmosphere. The reaction mixture was filtered through celite. The solvent removed and the residue was partitioned between EtOAc and sat. NaHCO3. The aqueous layer was extracted with EtOAc (3x 20 mL), the combined organic layers were washed with sat. NH4Cl, dried over Na2SO4 and the solvent evaporated. The residue was purified by column chromatography with 0-10% MeOH/DCM as eluent to give 1.2g of 14.3b (44% yield). MS analysis m/z = 237+1.
Preparation of N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethyl-butanamide (14T)
 14T
14T
[0273] Compound 14T was prepared from 14.2b and acid chloride a2 following general method A. MS analysis m/z - 335+1.
1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.43 (t, J= 7 Hz, 3H), 1.66 (q, J= 8 Hz, 2H), 2.53 (t, J= 4 Hz, 4H), 3.55 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 2H), 7.84 (s, 1H), 7.85 (br s, 1H), 8.26 (s, 1H).
[0274] Examples 14U - 14AD were prepared from 14.2b and the appropriate acid chlorides or acids following the procedure described for example 14T or 14L respectively.
14U: 1H NMR (CDCl3), δ 1.01 (s, 1H), 1.21 (s, 6H), 1.32 (s, 6H), 1.42 (t, J= 7 Hz, 3H), 2.50 (t, J= 5 Hz, 4H), 3.53 (s, 2H), 3.72 (t, J= 5 Hz, 4H), 4.08 (q, J= 7 Hz, 2H), 7.75 (br s, 1H), 7.83 (s, 1H), 8.13 (s, 1H).
14V: 1H NMR (CDCl3), δ 1.06 (s, 6H), 1.20 (s, 6H), 1.36 (s, 3H), 1.43 (t, J= 7 Hz, 3H), 2.53 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 4.08 (q, J= 7 Hz, 2H), 7.56 (br s, 1H), 7.82 (s, 1H), 8.23 (s, 1H).
14W: 1H NMR (CDCl3), δ 0.90 (t, J= 8 Hz, 3H), 1.23 (s, 3H), 1.43 (t, J= 7 Hz, 3H), 1.53 (m, 2H), 1.76 (m, 2H), 2.52 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.74 (t, 4H), 4.09 (q, 2H), 7.81 (br s, 1H), 7.84 (s, 1H), 8.26 (s, 1H).
14X: 1H NMR (CDCl3), δ 0.85 (t, J= 7 Hz, 9H), 1.43 (t, J= 7 Hz, 3H), 1.66 (q, J= 7 Hz, 6H), 2.52 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 2H), 7.84 (br s, 2H), 8.26 (s, 1H).
14Y: 1H NMR (CDCl3), δ 1.44 (t, J= 7 Hz, 3H), 1.76 (s, 3H), 2.53 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.76 (t, 4H), 4.11 (q, 2H), 7.88 (s, 1H), 8.20 (s, 1H), 8.24 (br s, 1H).
14Z: 1H NMR (CDCl3), δ 0.24 (m, 1H), 0.39 (m, 1H), 0.69 (m, 2H), 1.00 (m, 1H), 1.34 (d, J= 1 Hz, 3H), 1.43 (t, J= 7 Hz, 3H), 1.67 (m, 1H), 2.53 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.75 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 2H), 7.85 (s, 1H), 8.10 (br s, 1H), 8.29 (s, 1H).
14AA: 1H NMR (CDCl3), δ 0.49 (m, 2H), 0.63 (m, 2H), 1.08 (m, 1H), 1.13 (s, 6H), 1.43 (t, J= 7 Hz, 3H), 2.53 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 2H), 7.86 (s, 1H), 8.27 (s, 1H), 8.43 (br s, 1H).
14AB: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.43 (t, J= 7 Hz, 3H), 1.90 (m, 6H), 2.47 (m, 2H), 2.54 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.75 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 4H), 7.60 (br s, 1H), 7.83 (s, 1H), 8.29 (s, 1H).
14AC: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.43 (t, J= 7 Hz, 3H), 1.60 (m, 2H), 1.71 (m, 6H), 2.16 (m, 2H), 2.53 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 2H), 7.77 (br s, 1H), 7.83 (s, 1H), 8.25 (s, 1H).
14AD: 1H NMR (CDCl3), δ 0.87 (t, J= 8 Hz, 3H), 1.41 (m, 8H), 1.60 (m, 5H), 2.07 (m, 2H), 2.53 (t, J= 5 Hz, 4H), 3.55 (s, 2H), 3.74 (t, J= 5 Hz, 4H), 4.09 (q, J= 7 Hz, 2H), 7.84 (s, 1H), 7.86 (br s, 1H), 8.27 (s, 1H).
EXAMPLE 14AE:
Preparation of N-(5-(methylthio)-4-(morpholinomethyl)pyridin-2-yl)pivalamide (14AE)

[0275] A flask was charged with N-(5-Bromo-4-morpholin-4-ylmethylpyridin-2-yl)-2,2- dimethylpropionamide (2.4a3bl, 1.33 g, 3.72 mmol), N-methylpyrrolidinone (4 mL) and sodium methyl mercaptide (310 mg, 4.5 mmol). The reaction was heated at 130 °C for 16 hours. After cooling the reaction was diluted with 30 mL of water and 80 mL of ethyl acetate. The aqueous phase was back extracted with 80 mL of ethyl acetate. The combined organic extracts were washed with three 40 mL portions of water, dried with sodium sulfate and concentrated under vacuum. The crude reaction product was chromatographed 10% - 90% ethyl acetate/hexanes. (13% yield)
1H NMR (CD3OD), δ 1.32 (s, 9H), 2.55 (s, 3H), 3.40 (br s, 4H), 3.93 (br s, 4H), 4.55 (s, 2H), 8.28 (s, 1H), 8.50 (s, 1H).
[0276] Example 14AF was prepared from 14AE following the procedures described for example 14C.
1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 6H), 1.23 (s, 3H), 1.55 (m, 2H), 1.76 (m, 2H), 2.46 (s, 3H), 2.51 (t, J= 5 Hz, 4H), 3.57 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 7.91 (br s, 1H), 8.17 (s, 1H), 8.27 (s, 1H).
[0277] Example 14AG was prepared from 2A and sodium ethane thiolate and acid chloride a2 following the procedures described for example 14T.
1H NMR (CDCl3), δ 0.92 (t, J= 8 Hz, 3H), 1.26 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.67 (q, J= 7 Hz, 2H), 2.51 (t, J= 4 Hz, 4H), 2.88 (q, J= 7 Hz, 2H), 3.62 (s, 2H), 3.71 (t, J= 5 Hz, 4H), 8.04 (br s, 1H), 8.24 (s, 1H), 8.31 (s, 1H).
EXAMPLE 15A:
Preparation of methyl 6-amino-3-bromo^-methylisonicotinate (15.2)

[0278] Methyl 2-amino-6-methylisonicotinate (15.1, 3.5 g, 21.06 mmol) was dissolved in chloroform (200 mL). Bromine (1.11 mL, 21.6 mol) was slowly dripped in over 1 hour. Sodium thiosulfate solution (500 mL , 2M) was added to the reaction flask and it was left to stir for 15 minutes. The chloroform was separated off and the aqueous phase was extracted with chloroform (2 x 300 mL). The combined organic layers were evaporated. The crude product was purified via column chromatography using ethyl acetate/hexane as the eluent from 0-85% (19% yield). MS analysis m/z = 243+1.
Preparation of (6-amino-3-bromo-2-methylpyridin-4-yl)(morpholino)methanone (15.3)

[0279] Methyl 6-amino-3-bromo-2-methylisonicotinate (15.2, 0.6 g, 2.6 mmol) was dissolved in morpholine (bl, 20 mL). The reaction was heated to reflux and left to stir for 24 hours. The mixture was evaporated to dryness. To the residue was added morpholine (4 mL), and O- benzotriazol-1-yl-N,N,N',N'-tetramethyluronium tetrafluoroborate (TBTU, 1.334 g, 4.155 mmol) in acetonitrile (100 mL). N,N-diisopropylethylamine (1.36 mL, 7.79 mmol) was then dripped in and the reaction was left to stir for 24 hours. The solvent was evaporated off. The residue was taken up in sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated , dried over magnesium sulfate, and the solvent was evaporated (yield 88%). MS analysis m/z = 299+1.
Preparation of 5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-amine (15.4)
1 15.4

- I l l -
[0280] To (6-amino-3-bromo-2-methylpyridin-4-yl)(morpholino)methanone (15.3, 0.600 g, 2.00 mmol) in tetrahydrofiiran (20 mL) was added and borane-dimethyl sulfide complex (0.5 mL, 6 mmol). The reaction was heated to reflux for 3 hrs, then cooled to RT. Methanol (5ml) was added slowly and the mixture was stirred for lhr. Then 4M HCl in dioxane (5ml) was added and the reaction was heated to reflux for lhr. The mixture was concentrated under vacuum, diluted with sat. aq. sodium bicarbonate (20ml) and extracted with ethyl acetate (2x20ml). The organic layers were dried over MgSO4, the solvent was evaporated and the residue was purified via flash chromatography using 0-60% ethyl acetate in hexane as eluent (yield 92%). MS analysis m/z = 285+1.
Preparation of 5-methoxy-6-methyl-4-(morpholinomethyl)pyridin-2-amine (15.5)
15.5

[0281] In a microwave vial, 5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-amine (15.4, 0.360 g, 1.26 mmol), copper(I) bromide (0.361 g, 2.52 mmol), and sodium methoxide (3.15 g, 5.82 mmol, 25 wt.% in MeOH) were added together. The vial was sealed and evacuated. The vial was heated in the microwave at 100 °C for 90 minutes. The solution was filtered through a pad of celite, washing with ethyl acetate. The solvents were evaporated. The crude product was purified on a silica gel column using methanol/DCM from 0-30% as eluent (yield 33%). MS analysis m/z = 237 +1.
Preparation ofN-(5-methoxy-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2- dimethylbutanamide (15A)

[0282] Example 15A was prepared from 15.5 and 2,2-dimethylbutanoyl chloride a2 following general method A in 35 % yield. MS analysis m/z =335+1
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.60 (s, 3H), 1.66 (q, J= 7 Hz, 2H), 2.43 (s, 3H), 2.51 (br s, 4H), 3.50 (s, 2H), 3.72 (br s, 4H), 3.76 (s, 3H), 7.84 (br s, 1H), 8.11 (s, 1H).
EXAMPLE 16 A:
Preparation of 3-(methoxymethoxy)isonicotinaldehyde (16.2) B Li/DMF
 16.
16.
[0283] An oven dried flask was charged with the 3-(methoxymethoxy)pyridine (16.1, 10.6 g, 76.2 mmol), ether (200 mL) and NΛ,N,N-tetramethylethylenediamine (TMEDA, 12.6 mL, 83.8 mmol). The reaction was heterogeneous and was cooled to -70 °C with a 2-propanol/dry ice bath. N-butyllithium in hexane (1.6 M, 52 mL) was added dropwise over 30 minutes and the reaction was aged for 2.5 hours at this temperature. The DMF (8.2 mL, 110 mmol) was added dropwise over ten minutes and the reaction was allowed to slowly warm to room temperature and stir overnight. Most of the solid had settled out of suspension overnight. The reaction was cooled in an ice water bath and 20 mL of 7% citric acid was added dropwise over 30 minutes. After an hour an additional 60 mL of 7% citric acid was added. After 30 minutes of stirring the phases were allowed separate. The organic phase was dried with sodium sulfate and concentrated under vacuum. Yield was 8.5 g (47%) of a brown liquid.
Preparation of 4-((3-(methoxymethoxy)pyridin-4-yl)methyI)morpholine (16.3)

[0284] A flask was charged with the 3-(methoxymethoxy)isonictotinaldehyde (16.2, 7.5 g, 45 mmol) and tetrahydrofuran (50 mL). Morpholine (5.9 mL, 67 mmol) was added over two minutes and a precipitate rapidly began to form and the reaction warmed. The reaction was allowed to stir for 45 minutes and sodium triacetoxyborohydride (15 g, 72 mmol) was added in small portions over one hour. The reaction was allowed to stir at room temperature for 3 days. The reaction was cooled in an ice/water bath and 10 mL of methanol was added in portions over 90 minutes. The reaction was allowed to age for two hours and then concentrated under vacuum and partitioned between 125 mL of ethyl acetate and 80 mL of water. The aqueous phase was back extracted with 100 mL of ethyl acetate. The combined organic extracts were dried with sodium sulfate and concentrated under vacuum to yield 12.3 grams of crude product. 1.8 grams of this crude product was chromatographed on a silica column with a 2-propanol/ethyl acetate gradient up to 0 - 10%. MS analysis m/z = 238+1.
Preparation of 4-(morpholinomethyl)pyridin-3-ol (16.4)

[0285] To a solution of 4-((3-(methoxymethoxy)4-pyridin-4-yl)methyl)morpholine (16.3, 630 mg, 2.6 mmol) in methylene chloride (4 mL) was added hydrogen chloride in ether (6 mL, 2M) and a solid began to rapidly form. The reaction was allowed to stir overnight at room temperature. The reaction was concentrated under vacuum. The concentrate was cooled in an ice/water bath and a solution of 3 mL of concentrated ammonia in 5 mL of water was added followed by the addition of 30 mL of ethyl acetate. The aqueous phase was back extracted with 40 mL of ethyl acetate. The combined organic extracts were concentrated under vacuum to yield 300 mg of a solid. The aqueous phase was adjusted with 2 mL of saturated ammonium chloride and extracted with two 40 mL portions of ethyl acetate. An addtional 60 mg of solid was isolated and the solids were combined (yield 70%). MS analysis m/z = 194+1.
Preparation of 6-iodo-4-(morphoIinomethyl)pyridin-3-ol (16.5)

[0286] A flask was charged with the 4-(rnorpholinomethyl)pyridin-3-ol (16.4, 300 mg, 2 mmol), methanol (7 mL), sodium iodide (0.255 g, 1.70 mmol) and sodium hydroxide (0.0680 g, 1.70 mmol). The reaction was allowed to stir for 30 minutes at room temperature and the solids dissolved. The reaction was cooled in an ice/water bath and sodium hypochlorite in water (2.8 mL, 0.67 M) was added drop wise over three minutes. The reaction was aged for one hour. The reaction was cooled in an ice/water bath and 5 mL of 10% sodium bisulfite was added over 2 minutes. After ten minutes the reaction was diluted with seven mL of water and extracted with two 40 mL portions of ethyl acetate. The combined extracts were dried with sodium sulfate and concentrated under vacuum (yield 70%). MS analysis m/z = 320+1.
Preparation of 4-((2-iodo-5-propoxypyridin-4-yl)methyl)morpholine (16.6)

[0287] A flask was charged with the 6-iodo-4-(morpholinomethyl)pyridin-3-ol (16.5, 388 mg, 1.21 mmol), acetonitrile (12 mL), and potassium carbonate (760 mg, 5.5 mmol). 1-Iodopropane (190 μL, 1.9 mmol) was added and the reaction was heated at 70 °C overnight. The acetonitrile was removed under vaccum and ethyl acetate (50 mL) and water (30 mL) were added. The aqueous phase was back extracted with an addtional 50 mL of ethyl acetate. The combined extracts were dried with sodium sulfate and concentrated under vacuum to yield 16.6 as a brown liquid (yield 55%). MS analysis m/z = 362+1.
Preparation of 4-(morpholinomethyl)-5-propoxypyridin-2-amine (16.7)

[0288] A 5 mL microwave vial was charged with the t-butyl carbamate (141 mg, 1.21 mmol), xantphos (30 mg, 0.06 mmol) , cesium carbonate (441 mg, 1.35 mmol) and tris(dibenzylideneacetone)dipalladium (0) Pd2(dba)3 (20 mg, 0.02 mmol) and the vial was flushed with nitrogen for ten minutes. The 2-iodo-4-(morpholinomethyl)-5-propoxypyridine (16.6, 350 mg, 0.97 mmol) was dissolved in 1,4-dioxane (1.5 mL) and added as a solution. The reaction was flushed with nitrogen for an additional ten minutes and then heated at 70 °C overnight. The reaction was diluted with 30 mL of water and extracted with two 60 mL portions of ethyl acetate. The organic phase was dried with sodium sulfate and concentrated under vacuum. The material was purified via column chromatography using a 2-propanol/ ethyl acetate gradient from 0 to 100% (yield 20%). MS analysis m/z = 251+1.
Preparation of 3,3,3-trifluoro-2-methyl-Λ'-(4-(morphoIinomethyl)-5-propoxypyridin-2-yl)- 2-(trifiuoromethyl)propanamide (16A)

[0289] Compound 16A was prepared from 16.7 and acid el following the procedure described in example 14L.
1H NMR (CD3OD), δ 1.04 (t, J- 7 Hz, 3H), 1.79 (q, J= 7 Hz, 2H), 1.85 (s, 3H), 2.50 (t, J= 5 Hz, 4H), 3.63 (s, 2H), 3.69 (t, J= 5 Hz, 4H), 3.83 (t, J= 7 Hz, 2H), 7.51 (d, J= 5 Hz, 1H), 8.17 (d, J= 5 Hz, 1H).
EXAMPLE 17A:
Preparation ofN-(4-(hydroxymethyl)-5-methylpyridin-2-yl)-2,2-dimethyl-butanamide
(17.2)

[0290] To a solution of methyl 2-(2,2-dimethylbutanamido)-5-methylisonicotinate (17.1, 1.06 g, 4.00 mmol, obtained from methyl 2-amino-5-bromoisonicotinate and reactions according to general methods A and D) in THF (20 mL) under nitrogen atmosphere at 0 °C was added drop wise diisobutylaluminum hydride in tetrahydrofuran (14 mL, 1.0 M). The reaction mixture was stirred at 0 °C for 4 hours, quenched with sat. NH4Cl solution, filtered through a pad of celite, washed with EtOAc. The organic layer was separated, dried and concentrated in vacuo. The residue was purified by column chromatography using EtOAc-hexane (2:1) as eluent, yielding 600 mg of the product (yield 63%). MS analysis m/z = 236+1.
Preparation of Λ^^formyl-5-methylpyridin^-yl)-Σ^-dimethylbutanamide (17.3)

[0292] Compounds 17A - 17Z were prepared in a parallel fashion from aldehyde 17.3 and the amines shown in scheme 17 following this general procedure:

[0293] Solutions of aldehyde 17.3 (12 μmol/100 μL) and amines (10 μmol/100 μL) in methanol/ acetic acid (8:1) were prepared. Into a 96 well plate 100 μl of amine solution, 100 μL TMOF and 100 μL of aldehyde solution were combined and shaken over night at room temperature. PL-cyanoborohydride resin was added (3 eq.) and the plate was shaken overnight. The reaction mixtures were purified via SPE utilizing SCX-2 cartridges and methanol washes. The products were eluted with MeOH/NH3 (2.0M). MS analysis m/z results see table 1.
EXAMPLE 17AA:
Preparation ofN-(4-(1-hydroxypropyl)-5-methylpyridin-2-yl)-2,2-dimethyl-butanamide
(17.4)

[0294] To a solution of N-(4-formyl-5-methyl-pyridin-2-yl)-2,2-dimethyl-butyramide (17.3, 0.500 g, 2.13 mmol) in THF (40 mL) at -78 °C under nitrogen atmosphere was added ethylmagnesium bromide in tetrahydrofuran (6.4 mL, 1.0M). The reaction mixture was slowly warmed up to -30 °C over 2 hours. The reaction was quenched with sat. NH4Cl and extracted with EtOAC. The organic extracts were combined, dried and concentrated in vacuo. The
residue was purified by column chromatography using acetone-hexane (1 :3) as eluent, yielding 170 mg (30% yield). MS analysis m/z = 264+1.
Preparation of 2,2-dimethyl-N-(5-methyl-4-(1-morpholinopropyl)pyridiii-2-yl)butanamide
(17AA)

[0295] To a solution of 17.4 (0.150 g, 0.567 mmol) and triphenylphosphine (0.247 g, 0.942 mmol) in DCM (6 mL) at room temperature under nitrogen atmosphere was added carbon tetrabromide (0.312 g, 0.942 mmol) in portions. The reaction mixture was stirred at room temperature overnight, and then THF (6 mL), sodium bicarbonate (95.3 mg, 01.13 mmol) and morpholine (3.60 mL, 41.3 mmol) were added. The reaction mixture was stirred at 45 °C overnight, and then concentrated in vacuo. To the residue was added ether and 1 N HCl, the aqueous layer was separated and extracted with ether and then basified with 1 N NaOH, extracted with DCM. The organic extracts were combined, dried and concentrated in vacuo. The residue was purified by column chromatography using EtOAc-hexane (2:1) as eluent, yielding pure product (70 mg, 37% yield).
1H NMR (CDCl3), δ 0.74 (t, J= 8 Hz, 3H), 0.92 (X, J= I Hz, 3H), 1.28 (s, 6H), 1.67 (m, 4H), 2.29 (s, 3H), 2.37 (m, 2H), 2.53 (m, 2H), 3.42 (dd, J= 9 Hz and 4 Hz, 1H), 3.67 (m, 4H), 7.87 (br s, 1H), 8.02 (s, 1H), 8.19 (s, 1H).
EXAMPLE 18 A:
Preparation of 4-(benzyloxy)-2,3-dimethylpyridin-N-oxide (18.2)
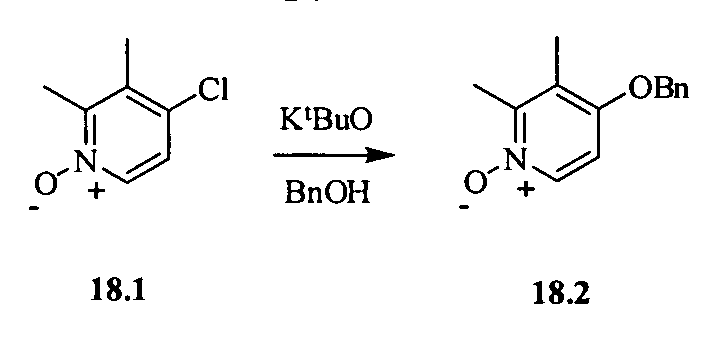
[0296] 4-Chloro-2,3-dimethylpyridine-N-oxide (18.1, 27.20 g, 0.1726 mol) was added to mixture of potassium tert-butoxide (25.00 g, 0.2228 mol) and benzyl alcohol (120 mL, 1.2 mol). The reaction mixture was heated at 120 °C for 8 days followed by evaporation of the benzyl alcohol under high vacuum. The residue was partitioned between water and DCM. The organic layer was separated, dried and concentrated in vacuo. The residue was purified by column
chromatography using acetone, DCM-MeOH (30:1 - 6:1) as eluent to yield the 4-benzyloxy substituted product 18.2 (13 g. 33%).
Preparation of 4-(benzyloxy)-6-chIoro-2,3-dimethylpyridine (18.3)

[0297] N-Oxide 18.2 (13.00 g, 0.05670 mol) was reacted with phosphoryl chloride (150 mL, 1.6 mol) at 90 - 95°C under N2 atmosphere for 2.5 hours. After evaporation of the excess POCl3, the residue was dissolved in DCM and washed with sat. NaHCO3. The organic layer was separated, dried and concentrated in vacuo. The residue was purified by column chromatography using EtOAc-hexane (1:4) as eluent, yielding 8.2 g of pure chloride 18.3 (yield 59%). MS analysis m/z = 247+1.
Preparation of ΛL(4-(benzyloxy)-5,6-dimethylpyridin-2-yi)-2,2-dimethylbutanamide (18.4)

[0298] In a round bottom flask, 18.3 (3.00 g, 12.1 mmol), xantphos (1.40 g, 2.42 mmol), palladium acetate (272 mg, 1.21 mmol), 1,4-dioxane (100 mL), potassium carbonate (4.18 g, 30.3 mmol), and 2,2-dimethyl-butyramide (3.49 g, 30.3 mmol) were added and the reaction was heated at 100 °C for 24 hours under a nitrogen atmosphere.
The reaction slurry was filtered through celite, washing with DCM. The solvents were evaporated. The residue was purified by silica gel column chromatography using ethyl acetate- hexane (2:3) as eluent, yielding 3.1 g of the desired coupling product, 18.4 (78% yield). MS analysis m/z = 326+1.
Preparation of N-(4-hydroxy-5,6-dimethyIpyridin-2-yl)-2,2-dimethyIbutanamide (18.5)

Preparation of 6-(2,2-dimethylbutanamido)-2,3-dimethylpyridin-4-yl trifluoromethanesulfonate (18.6)

[0300] To a solution of 18.5 (1.00 g, 4.23 mmol) in methylene chloride (60 mL) was added triethylamine (1.47 mL, 10.6 mmol), 4-dimethylaminopyridine (77.5 mg, 0.635 mmol), followed by N-phenylbis(trifluoromethanesulphonimide) (2.12 g, 5.92 mmol). The reaction mixture was stirred at room temperature overnight, then washed with aqueous sat. NaHCO3, dried over Na2SO4, and concentrated in vacuo. The residue was chromatographed using EtOAc:hexane (1 :2) as eluent, yielding the inflate 18.6 (98% yield). MS analysis m/z = 368+1.
Preparation ofN-(4-(4-fluorophenethyl)-5,6-dimethylpyridin-2-yl)-2,2-di- methylbutanamide (18A)

[0301] To a solution of triflate 18.6 (0.300 g, 0.814 mmol) in THF (10 mL) at room temperature was added tetrakis(triphenylphosphine)palladium(0) (94.1 mg, 0.0814 mmol) followed by drop wise addition of 0.50 M of 4-fluorophenethylzinc bromide in tetrahydrofuran (9.8 mL). The reaction was stirred at 55 °C overnight. The reaction was quenched with sat. NH4Cl and extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was evaporated and the residue was purified by column
chromatography using ethyl acetate-hexane (1:2) as eluent, yielding the desired product (180 mg, 65% yield).
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.29 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 2.13 (s, 3H), 2.41 (s, 3H), 2.85 (s, 4H), 6.97 (m, 2H), 7.14 (m, 2H), 7.81 (br s, 1H), 8.00 (s, 1H)
[0302] Compounds 18B and 18C were prepared from triflate 18.6 following the procedure described in example 18A utilizing pyridylzinc bromide and benzylzinc bromide respectively.
18B: 1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.21 (s, 3H), 2.49 (s, 3H), 7.29 (m, 1H), 7.45 (d, J= 8 Hz, 1H), 7.76 (m, 1H), 7.89 (br s, 1H), 8.14 (s, 1H), 8.69 (dd, J= 5 Hz and 1 Hz, 1H)
18C: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.09 (s, 3H), 2.40 (s, 3H), 3.98 (s, 2H), 7.13 (m, 2H), 7.17 (m, 1H), 7.26 (m, 2H), 7.81 (br s, 1H), 8.04 (s, 1H)
EXAMPLE 18D
Preparation of methyl 6-(2,2-dimethylbutanamido)-2,3-dimethylisonicotinate (18.7)

[0303] To the solution of triflate 18.6 (1.56 g, 4.23 mol) in DMF (15 mL) was added methanol (8 mL), triethylamine (1.2 mL, 8.5 mmol), 1,3-bis(diphenylphosphino)propane (140 mg, 0.339 mmol) followed by palladium acetate (76.1 mg, 0.339 mmol). The reaction mixture was heated to 65 °C and carbon monoxide was bubbled through the reaction solution for 3.5 hours. The reaction mixture was then cooled to room temperature and diluted with ether, washed with water, brine, dried and concentrated in vacuo. The residue was purified by column chromatograph using EtOAc:hexane (1:2) as eluent to give the methyl ester 18.7 (990 mg, 84% yield). MS analysis m/τ = 278+1.
Preparation of N-[4-(1,1-Dioxo-llambda *6*-thiomorpholin-4-ylmethyl)-5,6-dimethyl- pyridin-2-yl]-2,2-dimethyl-butyramide (18D)
 18D
18D
[0304] Compound 18D was prepared from ester 18.7 following procedures as described for the synthesis of compounds 17.2 and 17AA.
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.21 (s, 3H), 2.44 (s, 3H), 3.01 (m, 4H), 3.08 (m, 4H), 3.59 (s, 2H), 7.84 (br s, 1H), 8.05 (s, 1H)
EXAMPLE 19 A:
Preparation of 4-phenylpyridin-2-amine (19.2)

[0305] Compound 19.2 was prepared from 4-bromopyridin-2-amine 19.1 and phenylboronic acid in 64% yield following general method C. MS analysis m/z = 170+1.
Preparation of 5-bromo-4-phenylpyridin-2-amine (19.3)

[0306] Compound 19.3 was prepared from 19.2 in 12% yield following the procedure described for the synthesis of compound 15.2 in example 15A. MS analysis m/z = 247+1.
Preparation of yV-(5-bromo-4-phenylpyridin-2-yl)-2,2-dimethylbutanamide (19A)
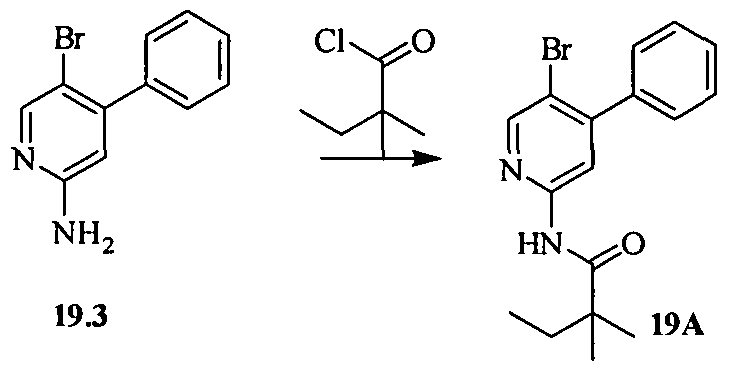
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 7.45 (m, 5H), 7.99 (br s, 1H), 8.34 (s, 1H), 8.45 (s, 1H).
EXAMPLE 19B;
Preparation of 5-methoxy-4-phenylpyridin-2-amine (19.4)

[0308] In a microwave vial, 5-bromo-4-phenyl-pyridin-2-ylamine (19.3, 0.900 g, 3.61 mmol) and copper (0.5740 g, 9.032 mmol) were suspended in sodium methoxide (6.04 mL, 25 wt% in MeOH). The vial was evacuated and run for 3 hours at 150 °C.
The reaction was filtered through a pad of celite, washing with ethyl acetate. The solution was washed with sat. ammonium chloride, brine, and dried over magnesium sulfate. The crude product was purified on a silica gel column using an acetone/hexane gradient (10-75%, yield 44%). MS analysis m/z = 200+1.
Preparation ofN-(5-methoxy-4-phenylpyridin-2-yl)-2,2-dimethylbutanamide (19B)

[0309] Compound 19B was prepared from 19.4 and acid chloride a3 in 58% yield following general procedure A.
1H NMR (CDCl3), δ 0.92 (t, J= 7 Hz, 3H), 1.29 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 3.87 (s, 3H), 7.41 (m, 3H), 7.62 (m, 2H), 7.91 (br s, 1H), 7.98 (s, 1H), 8.32 (s, 1H).
EXAMPLE 20A:
Preparation of 2-chloro-6,7-dihydro-5H-cycIopenta[b]pyridine-4-carboxylic acid (20.1)
10 20.1
 [0310] In a round bottom flask, compound 10.3dl (10.0 g, 55.812 mmol) was dissolved in phosphoryl chloride (50 mL). The solution was heated to 90 0C and left to stir for 1 hour. The flask was cooled to room temperature and then cooled further in an ice bath. A solution of sodium bicarbonate was SLOWLY dripped into the flask over 40 minutes until the pH was approximately 8-9. The solution was extracted with ethyl acetate. TLC of the organic layer showed no UV absorbance. TLC of the aqueous layer showed that the product was still in the aqueous layer, most likely as the carboxyl salt. The ethyl acetate was separated off and 6.0 M aqueous HCl was dripped into the aqueous layer until a pH of approximately 4-5 was reached. Ethyl acetate was added once again, this time TLC of the organic layer showed UV absorbance. The aqueous layer was extracted 3 times with ethyl acetate, the organics were separated off, washed with brine, dried over magnesium sulfate, and then concentrated down to give 20.1 as a reddish white color solid (yield 20%). MS analysis m/z = 278+1.
[0310] In a round bottom flask, compound 10.3dl (10.0 g, 55.812 mmol) was dissolved in phosphoryl chloride (50 mL). The solution was heated to 90 0C and left to stir for 1 hour. The flask was cooled to room temperature and then cooled further in an ice bath. A solution of sodium bicarbonate was SLOWLY dripped into the flask over 40 minutes until the pH was approximately 8-9. The solution was extracted with ethyl acetate. TLC of the organic layer showed no UV absorbance. TLC of the aqueous layer showed that the product was still in the aqueous layer, most likely as the carboxyl salt. The ethyl acetate was separated off and 6.0 M aqueous HCl was dripped into the aqueous layer until a pH of approximately 4-5 was reached. Ethyl acetate was added once again, this time TLC of the organic layer showed UV absorbance. The aqueous layer was extracted 3 times with ethyl acetate, the organics were separated off, washed with brine, dried over magnesium sulfate, and then concentrated down to give 20.1 as a reddish white color solid (yield 20%). MS analysis m/z = 278+1.
Preparation of methyl 2-(2,2-dimethylbutanamido)-6,7-dihydro-5H-cyclopenta- [b]pyridine-4-carboxylate (20.2)

[0311] Compound 20.2 was prepared from the methyl ester of 20.1 and 2-cyclopropyl-2- methylpropanamide following the procedure described for the synthesis of 18.4 in example 18 A. MS analysis m/z = 302+1.
Preparation of 2-cyclopropyl-7V-[4-(l,l-dioxo-llambda*6*-thio-morpholin-4-ylmethyl)-6,7- dihydro-5H-[l]pyrindin-2-yl]-isobutyramide (20A)
2OA

[0312] Compound 20A was prepared from ester 20.2 following procedures as described for the synthesis of compounds 17.2 and 17AA.
1H NMR (CDCl3), δ 0.49 (m, 2H), 0.63 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 2.13 (m, 2H), 2.87 (t, J= 7 Hz, 2H), 2.94 (t, J- 8 Hz, 2H), 3.01 (m, 4H), 3.10 (m, 4H), 3.59 (s, 2H), 8.08 (s, 1H), 8.44 (br s, 1H).
EXAMPLE 21A:
Preparation of diethyl (5-bromo-2-(2,2-dimethylbutanamido)pyridin-4-yl)methyl- phosphonate (21.1a)
Toluene
 2_
2_
[0313] To a flask at room temperature containing N-(5-bromo-4-bromomethylpyridin-2-yl)- 2,2-dimethylbutanamide (2.3a2, 2.00 g, 5.49 mmol) under an atmosphere of nitrogen was added anhydrous toluene (11 mL) with stirring, to give a clear, colorless solution. Triethyl phosphite (0.5 mL, 3 mmol) was then added drop wise, and the reaction was heated at an oil bath temperature of 105 °C overnight.
The crude reaction was concentrated and the residue was purified via column chromatography using a gradient from 0 % EtOAc/hexanes to 100% EtOAc/hexanes (0.71 g 21.1a, 30% yield). . MS analysis w/z = 420+1.
Preparation ofN-(4-((1-acetylpiperidin-4-ylidene)methyl)-5-bromopyridin-2-yl)-2,2- dimethylbutanamide (21.2a)

[0314] To a flask containing a solution of diethyl (5-bromo-2-(2,2- dimethylbutanamido)pyridin-4-yl)methylphosphonate (21.1a, 0.35 g, 0.83 mmol) in anhydrous tetrahydrofuran (7.96 mL) under an atmosphere of nitrogen at -78 °C in a dry ice/acetone bath was added drop wise potassium tert-butoxide (0.91 mmol, 0.91 mL of a 1.0 M solution in THF), and the resulting light yellow-orange solution was stirred for ~ 2 hours at this temperature. A clear solution of 1-acetyl-4-piperidone (0.12 mL, 1.0 mmol) in anhydrous tetrahydrofuran (1.6 mL) was then added drop wise. The reaction was allowed to warm to room temperature
overnight. The reaction was then heated at 55 °C for 2 hours and subsequently allowed to cool to room temperature. The reaction was partitioned between saturated aqueous NaHCO3 and EtOAc. The layers were separated, and the aqueous phase was extracted once more with EtOAc. The combined organic layer was dried over Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography using a gradient from 0 % EtOAc/hexanes to 100% EtOAc yielding 213.4 mg of product 21.2a (63% yield). MS analysis m/z = 407+1.
Preparation ofN-(4-((1-acetylpiperidin-4-ylidene)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide (21.3a)

[0315] Compound 21.3a was obtained from 21.2a in 82% yield following general method D. MS analysis m/z = 343+1.
Preparation ofN-(4-((1-acetylpiperidin-4-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide (21A)
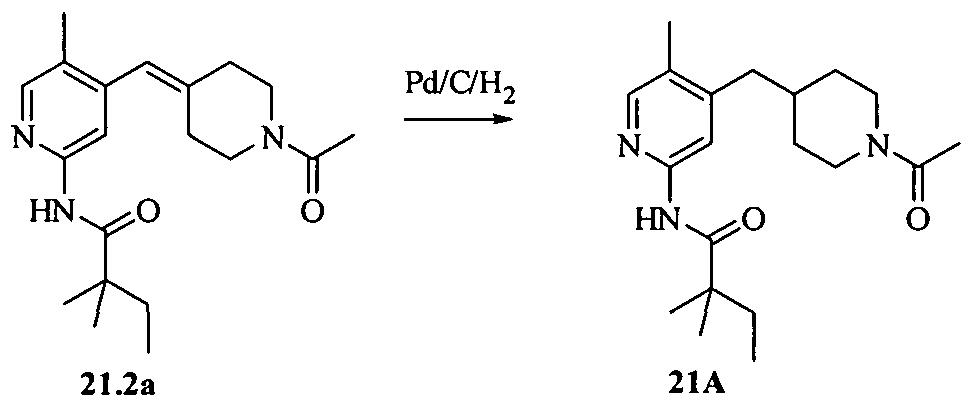
[0316] A flask containing a stir bar and 7V-(4-((1-acetylpiperidin-4-ylidene)methyl)-5- methylpyridin-2-yl)-2,2-dimethylbutanamide (21.2a, 103 mg, 0.3mmol) as a solution in anhydrous methanol (7.0 mL, 0.17 mol) was evacuated and purged with nitrogen several times before the addition of palladium on carbon (10 mg, 0.08 mmol). The flask was then purged and evacuated with hydrogen (100 mL, 6 mmol). The reaction was allowed to stir overnight under a balloon of hydrogen. The mixture was filtered through celite under vacuum with a MeOH rinse and concentrated. The crude product was purified by column chromatography using an EtOAc/hexane gradient 0-100% yielding 61.0 mg of product 21A (59% yield).
1H NMR (CDCl3), δ 0.92 (t, J= 7 Hz, 3H), 1.23 (m, 2H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 1.72 (m, 1H), 1.83 (m, 1H), 2.08 (s, 3H), 2.23 (s, 3H), 2.48 (m, 2H), 2.59 (m, 2H), 2.97 (dt, J= 12 Hz and 3 Hz, 1H), 3.79 (m, 1H), 4.62 (m, 1H), 7.89 (br s, 1H), 8.01 (s, 1H), 8.05 (s, 1H).
[0317] Example 21B was obtained starting from compound 20.4 following the procedures described in example 21 A.
1H NMR (CDCl3), δ 0.48 (m, 2H), 0.62 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 1.19 (m, 1H), 1.27 (m, 1H), 1.68 (m, 3H), 2.08 (s, 3H), 2.11 (m, 2H), 2.49 (m, 3H), 2.83 (t, J= 7 Hz, 2H), 2.94 (t, J = 8 Hz, 2H), 2.99 (m, 1H), 3.78 (m, 1H), 4.60 (m, 1H), 7.90 (br s, 1H), 8.43 (s, 1H)
[0318] Examples 21C - 21D were obtained from 2.3a2 and dihydro-2H-pyran-4(3H)-one and dihydro-2H-thiopyran-4(3H)-one (oxidation to the sulfone was achieved with peroxyacetic acid) following the procedures described in example 21 A.
21C: 1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.40 (m, 2H), 1.57 (dd, J= 13 Hz and 2 Hz, 2H), 1.66 (q, J= 7 Hz, 2H), 2.24 (s, 3H), 2.55 (d, J= 7 Hz, 2H), 3.34 (m, 2H), 3.95 (dd, J= 12 Hz and 4 Hz, 2H), 7.90 (br s, 1H), 8.01 (s, 1H), 8.05 (s, 1H).
21D: 1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.28 (s, 6H), 1.66 (q, J= 7 Hz, 2H), 1.93 (m, 3H), 2.05 (m, 2H), 2.23 (s, 3H), 2.61 (d, J= 6 Hz, 2H), 2.94 (m, 2H), 3.04 (m, 2H), 7.91 (br s, 1H), 8.04 (s, 1H), 8.08 (s, 1H).
EXAMPLE 22A
Preparation of 1-(3-Hydroxypyridin-2-yl)-Λ',Λ',Λ'-trimethyl-methanaminiuni iodide (22.2)

[0319] A solution of 2-(dimethylaminomethyl)-3-hydroxypyridine (22.1, 10.64 g, 0.06991 mol) in acetone (30 mL) was cooled to 0 °C. To this was added, drop wise, methyl iodide (4.36 mL, 0.0700 mol) and the mixture warmed to room temperature overnight. A precipitate formed, which was collected under filtration to give the product as a pale yellow solid (20 g, 100%).
1H NMR (DMSO), δ 3.22 (s, 9H), 4.61 (s, 2H), 7.40 (t, J= 2 Hz, 2H), 8.21 (dd, J= 2 Hz and 2 Hz, 1H). MS analysis m/z = 166+1.
Preparation of 2,3-Dihydrofuro[3,2-b]pyridine (22.3)
 223
223
[0320] To a solution of trimethylsulfoxonium iodide (7.6 g, 0.035 mol) in DMSO (42 mL) was added sodium hydride (2 g, 0.07 mol) and the mixture stirred at room temperature for 1 hour. A solution of (3-hydroxypyridin-2-ylmethyl)trimethylammonium iodide (22.2, 10 g, 0.03 mol) in DMSO (30 mL) was added and the mixture stirred at room temperature overnight. Monitoring of the reaction using LCMS indicated a new, more lipophilic product (no ionization). The mixture was poured into ice water and extracted with chloroform. The organic extracts were washed with brine, dried over sodium sulfate, filtered and concentrated under reduced pressure. Column chromatography on silica using an ethyl acetate/hexanes solvent system afforded the desired product as a white solid (3.06 g, 70%).
1H NMR (CDCl3), δ 3.34 (t, J= 9 Hz, 2H), 4.66 (t, J= 9 Hz, 2H), 7.01 (d, J= 3 Hz, 2H), 8.04 (t, J= 3 Hz, 1H)
Preparation of 5-Nitro-2,3-dihydrofuro[3,2-b] pyridine (22.4)

[0321] To a solution of 2,3-dihydrofuro[3,2-b]pyridine (22.3, 3.82 g, 0.0315 mol) in sulfuric acid (20 mL) cooled to 0 °C was added, drop wise, a mixture of nitric acid (3 mL, 0.07 mol) and sulfuric acid (3 mL) and the mixture stirred at 0 °C for 1 hour. Thin layer chromatography indicated complete reaction. The reaction mixture was poured into ice water and extracted with chloroform. The organic extracts were washed with water, brine, dried over sodium sulfate, filtered and evaporated under reduced pressure to give the product as a white solid (4.91 g, 93%).
1H NMR (CDCl3), δ 3.46 (t, J= 9 Hz, 2H), 4.87 (t, J= 9 Hz, 2H), 7.18 (d, J= 9 Hz, 1H), 8.15 (d, J= 9 Hz, 1H). MS analysis m/z = 166+1.
Preparation of 2,3-Dihydrofuro[3,2-Λ]pyridin-5-ylamine (22.5)

1H NMR (CDCl3), δ 3.18 (t, J= 8 Hz, 2H), 4.12 (br s, 2H), 4.57 (t, J= 9 Hz, 2H), 6.27 (d, J= 9 Hz, 1H), 6.90 (d, J= 9 Hz, 1H)
Preparation of N-(2,3-Dihydrofuro[3,2-A]pyridin-5-yl)-2,2-dimethylbutyramide (22.6)

[0323] To a solution of 2,3-dihydrofuro[3,2-b]pyridin-5-ylamine (22.5, 1.00 g, 0.00734 mol) in methylene chloride (20 mL) was added triethylamine (3.1 mL, 0.022 mol) and the mixture was cooled to 0 °C before 2,2-dimethylbutanoyl chloride (a2, 1.2 mL, 0.0088 mol) was added. The mixture was allowed to warm to room temperature over 1 hour. The reaction mixture was poured into water and extracted with methylene chloride. The organic extracts were washed with brine, dried over sodium sulfate, filtered and evaporated under reduced pressure. The crude material was passed through a plug of silica, eluting with 50% ethyl acetate in hexanes to give the product as a pale yellow solid (1.72 g, 100%).
1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.27 (s, 6H), 1.65 (q, J= 8 Hz, 2H), 3.25 (t, J= 9 Hz, 2H), 4.65 (t, J= 9 Hz, 2H), 7.06 (d, J= 9 Hz, 1H), 7.84 (br s, 1H), 8.02 (d, J= 9 Hz, 1H). MS analysis m/z - 234+1.
Preparation of 2,2-Dimethyl-N-(4-oxy-2,3-dihydrofuro[3,2-Λ]pyridin-5-yl)-butyrainide
(22.7)

[0324] To a solution of JN-(2,3-dihydrofuro[3,2-b]pyridin-5-yl)-2,2-dimethylbutyramide (22.6, 1.22 g, 0.00521 mol) in chloroform (25 mL) was added w-chloroperbenzoic acid (1.08 g, 0.00625 mol) and the mixture stirred at room temperature over the weekend. The reaction
mixture was washed twice with saturated sodium bicarbonate solution, water, brine, dried over sodium sulfate, filtered and evaporated to give the product as a yellow solid (1.30 g, 99%).
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.31 (s, 6H), 1.69 (q, J= 7 Hz, 2H), 3.51 (t, J= 9 Hz, 2H), 4.74 (t, J= 9 Hz, 2H), 6.81 (d, J= 9 Hz, 1H), 8.27 (d, J= 9 Hz, 1H), 10.03 (br s, 1H). MS analysis m/z = 250+1.
Preparation of7V-(7-Bromo-4-oxy-2,3-dihydrofuro[3,2-A]pyridin-5-yl)-2,2-dimethyl- butyramide (22.8)

[0325] To a solution of 2,2-dimethyl-N-(4-oxy-2,3-dihydrofuro[3,2-b]pyridin-5-yl)butyramide (22.7, 200 mg, 0.0008 mol) in carbon tetrachloride (5 mL) was added bromine (0.045 mL, 0.00088 mol) and the reaction mixture stirred at room temperature over the weekend. The reaction mixture was poured into saturated sodium thiosulfate solution and extracted with ethyl acetate. The organic extracts were washed with brine, dried over sodium sulfate, filtered and evaporated under reduced pressure. Column chromatography on silica using an ethyl acetate/hexanes solvent system afforded the desired product as a pale yellow solid (70 mg, 30%).
1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.30 (s, 6H), 1.69 (q, J= 8 Hz, 2H), 3.60 (t, J= 9 Hz, 2H), 4.82 (t, J= 9 Hz, 2H), 8.56 (s, 1H), 9.96 (br s, 1H). MS analysis m/z = 328+1.
Preparation of 2,2-Dimethyl-N-(4-oxy-7-viny--2,3-dihydrofuro[3,2-b]pyridin-5- yl)butyramide (22.9)

[0326] To a solution of N-(7-bromo-4-oxy-2,3-dihydrofuro[3,2-b]pyridin-5-yl)-2,2- dimethylbutyramide (22.8, 620 mg, 0.0019 mol) in 1,4-dioxane (10 mL) and water (3 mL) was added potassium carbonate (781 mg, 0.00565 mol), 4,4,5, 5 -tetramethyl-2-vinyl- 1,3,2- dioxaborolane (0.351 mL, 0.00207 mol) and tetrakis(triphenylphosphine) palladium(0) (220 mg, 0.00019 mol) and the mixture was heated at 110 °C for 2 hours. The reaction mixture was concentrated in vacuo. Column chromatography on silica using an ethyl acetate/hexanes solvent system afforded the desired product as a pale yellow solid (410 mg, 79%).
1H NMR (CDCl3), δ 0.91 (t, J= 8 Hz, 3H), 1.24 (s, 6H), 1.70 (q, J= 8 Hz, 2H), 3.51 (t, J= 9 Hz, 2H), 4.79 (t, J= 9 Hz, 2H), 5.53 (dd, J= 11 Hz and 1 Hz, 1H), 6.10 (dd, J= 18 Hz and 1 Hz, 1H), 6.60 (dd, J= 18 Hz and 12 Hz, 1H), 8.33 (s, 1H), 10.01 (br s, 1H). MS analysis m/z = 276+1.
Preparation of N-(7-FormyI-4-oxy-2,3-dihydrofuro [3,2-b] pyridin-5-yI)-2,2-di- methylbutyramide (22.10)

[0327] To a solution of 2,2-dimethyl-N-(4-oxy-7-vinyl-2,3-dihydrofuro[3,2-b]pyridin-5- yl)butyramide (22.9, 40 mg, 0.0001 mol) in tetrahydrofuran (3 mL) and water (1 mL) was added 4 M of N-methylmorpholine N-oxide in water (0.07 mL) and 0.1 M of osmium tetraoxide in tert- butyl alcohol (0.2 mL) and the reaction stirred at room temperature for 3 hours. LCMS indicated complete diol formation. To this was then added sodium periodate (69.7 mg, 0.000326 mol) and the reaction continued for a further 2 hours. LCMS indicated complete conversion to the aldehyde. The reaction mixture was poured into 10% sodium thiosulfate solution and extracted with ethyl acetate. The organic extracts were washed with brine, dried over sodium sulfate, filtered and evaporated under reduced pressure to give the product as a pale yellow solid (40 mg, 100%).
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.32 (s, 6H), 1.70 (q, J= 8 Hz, 2H), 3.53 (t, J= 8 Hz, 2H), 4.92 (t, J= 9 Hz, 2H), 8.69 (s, 1H), 9.83 (br s, 1H), 10.06 (s, 1H). MS analysis m/z = 278+1.
Preparation of 2,2-Dimethyl-N-(7-morpholin-4-ylmethyl-4-oxy-2,3-dihydrofuro[3,2- 6]pyridin-5-yl)butyramide (22.11)

[0328] To a solution of N-(7-formyl-4-oxy-2,3-dihydrofuro[3,2-b]pyridin-5-yl)-2,2-dimethyl- butyramide (22.10, 40 mg, 0.0001 mol) in 1,2-dichloroethane (2 mL) was added morpholine (0.0251 mL, 0.000287 mol) and the mixture cooled in an ice bath. Sodium triacetoxyborohydride (76 mg, 0.00036 mol) was added and the mixture warmed to room temperature overnight. LCMS indicated complete reaction. Saturated sodium bicarbonate
solution was added and the mixture stirred for 30 minutes. The layers were separated and the aqueous portion was extracted with methylene chloride. The organic extracts were washed with brine, dried over sodium sulfate, filtered and evaporated under reduced pressure. Column chromatography on silica using a methanol/methylene chloride solvent system afforded the desired product as a brown oil (20 mg, 40%).
1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.31 (s, 6H), 1.70 (q, J= 7 Hz, 2H), 2.49 (t, J= 5 Hz, 4H), 3.51 (t, J= 9 Hz, 2H), 3.72 (t, J= 5 Hz, 4H), 4.74 (t, J= 9 Hz, 2H), 8.31 (s, 1H), 10.04 (br s, 1H). MS analysis rø/z = 349+1.
Preparation of 2,2-Dimethyl-N-(7-morpholin-4-ylmethyl-2,3-dihydrofuro[3,2-b]pyridin-5- yl)butyramide (22A)

[0329] To a solution of 2,2-dimethyl-N-(7-morpholin-4-ylmethyl-4-oxy-2,3-dihydrofuro[3,2- 6]pyridin-5-yl)butyramide (22.11, 20 mg, 0.00006 mol) in ethanol (3 mL) was added Raney nickel (3 mg, 0.00006 mol) and the mixture stirred under an atmosphere of hydrogen overnight. LCMS indicated complete reaction. The mixture was filtered through celite, washing with ethanol and the filtrate was evaporated under reduced pressure. Column chromatography on silica using a methanol/methylene chloride solvent system afforded the desired product as a white solid (1 mg, 5%).
1H NMR (CDCl3), δ 0.90 (t, J= 7 Hz, 3H), 1.23 (t, J= 8 Hz, 2H), 1.27 (s, 6H), 1.66 (q, J= 8 Hz, 2H), 2.57 (br s, 4H), 2.74 (t, J= 8 Hz, 2H), 3.76 (br s, 4H), 7.80 (br s, 1H), 7.85 (s, 1H). MS analysis m/τ = 333+1.
EXAMPLE 23A:
Preparation of N-(5-bromo-4-(cyanomethyl)pyridin-2-yl)-2,2-dimethylbutanamide (23.1)

[0330] A flask charged with the N-(5-bromo-4-bromomethylpyridin-2-yl)-2,2-dimethyl- butanamide (2.3a2, 2.45 g, 0.00673 mol) in acetonitrile (25 mL) and stirred for 20 minutes. Water (10 mL) was added and the reaction became cloudy and potassium cyanide (1200 mg,
0.018 mol) was added and the reaction was allowed to stir overnight at room temperature. The reaction was concentrated under vacuum and the residue was partitioned between 50 mL of water and 120 mL of ethyl acetate. The aqueous phase was back extracted with an additional 80 mL of ethyl acetate. The combined organic extracts were dried with sodium sulfate and concentrated under vacuum. The concentrate was chromatographed on silica gel with an ethyl acetate/hexane gradient (5 to 60%.) yielding 346 mg (17%). MS analysis m/z = 309+1.
Preparation of N-(4-(cy anomethyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide (23.2)

[0331] A microwave vial was charged with the potassium phosphate (931 mg, 0.00438 mol), methylboronic acid (262 mg, 0.00438 mol) and a fresh bottle of [1,1'-bis(diphenyl- phosphino)ferrocene]dichloropalladium(II),complex with dichloromethane (1:1) (90 mg, 0.00011 mol) and the vial was swept with nitrogen for 15 minutes. N-(5-bromo-4- (cyanomethyl)pyridin-2-yl)-2,2-dimethylbutanamide (23.1, 340 mg, 0.0011 mol) dissolved in 1,4-dioxane (5 mL, 0.07 mol) was added via syringe followed by the addition of water (1.00 mL, 0.0555 mol). The reaction was microwave heated at 145 °C for 80 minutes. The reaction was diluted with 25 mL of water and 100 mL of ethyl acetate. The organic phase was filtered through celite, dried with sodium sulfate and concentrated under vacuum. The residue was chromatographed on a silica gel column and eluted with ethyl acetate/hexanes: 15% ethyl acetate/hexanes to 100% ethyl acetate (Yield 190 mg, 53%). MS analysis m/z = 245+1.
Preparation of 7V-(4-(aminoethyl)-5-methyIpyridin-2-yl)-2,2-dimethylbutnamide (23.3)

[0332] A Parr reaction flask was charged with the N-(4-(cyanomethyl)-5-methylpyridin-2-yl)- 2,2-dimethylbutanamide (23.2, 191 mg, 0.000778 mol), ethanol (8 mL), ammonium hydroxide (5
mL, 0.1 mol) and rhodium (150 mg, 0.0014 mol) on alumina (10%). The reaction was hydrogenated on a Parr shaker at 50 psi overnight. The next day the reaction was purged with nitrogen. The reaction was filtered through celite and concentrated under vacuum (yield 140 mg, 65%). MS analysis m/z = 249+1.
Preparation of 2,2-dimethyI-7V-(5-methyl-4-(2-morpholinoethyl)pyridin-2-yl)-butanamide
(23A)

[0333] To a flask charged with N-(4-(aminoethyl)-5-methylpyridin-2-yl)-2,2- dimethylbutnamide (23.3, 120.0 mg, 0.0004812 mol), acetonitrile (500 uL, 0.01 mol) and potassium carbonate (190 mg, 0.0014 mol), bis(2-bromomethyl)ether (60.0 uL, 0.000477 mol) was added and the reaction was heated at 70 °C in an oil bath over the weekend. After cooling the reaction was diluted with 30 mL of ethyl acetate and 15 mL of water. The aqueous phase was back extracted with an additional 30 mL of ethyl acetate. The combined organic extracts were dried with sodium sulfate and concentrated under vacuum. The residue was purified by prep. HPLC (yield: 102 mg, 64%).
1H NMR (CDCl3), δ 0.91 (br s, 3H), 1.30 (s, 6H), 1.70 (br s, 2H), 2.31 (br s, 3H), 3.17 (m, 6H), 4.02 (m, 6H), 8.02 (s, 1H), 8.25 (s, 1H), 9.33 (br s, 1H).
EXAMPLE 24A
Preparation of 4-chIoro-3-methylpyridine 1 -oxide (24.2a)

[0334] 3-Methyl-4-nitropyridine 1-oxide (24.1a, 12.0 g, 0.07786 mol) was dissolved in acetyl chloride (60.00 mL). The reaction was heated to reflux for 2 hours. After cooling the reaction was poured onto ice and was basified with anhydrous sodium carbonate and extracted with chloroform. The extract was dried with potassium carbonate and filtered. The chloroform was evaporated off to yield the desired product in high purity. (85% yield) The MP was taken and found to be at 124 °C, which matches literature references.
Preparation of 4-mercapto-3-methylpyridine 1-oxide (24.3a)
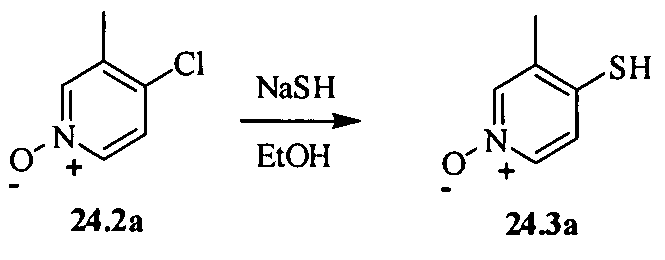
[0335] 4-Chloro-3-methylpyridine 1-oxide (24.2a, 13.74 g, 0.09570 mol) and sodium hydrogen sulfide (15.39 g, 0.2745 mol) were dissolved in ethanol (800 mL). The reaction was left to stir at reflux for 1 week. The solution was filtered, the solvent evaporated, and the residue washed with ether, yielding 24.3a as a solid quantitatively.
Preparation of 3-methyl-4-(morpholinosulfonyl)pyridine 1-oxide (24.4a)

[0336] 4-Mercapto-3-methylpyridine 1-oxide (24.3a, 14.0 g, 0.099156 mol) was stirred in a solution of 9.0 N hydrogen chloride (163.92 mL, 5.3500 mol). The reaction was cooled to -5 °C. Chlorine gas was then bubbled in and the reaction was left to stir for 1.5 hours, maintaining the temperature at -5 °C.
Calcium carbonate (12.61 g, 0.1260 mol) was added slowly while keeping the temperature at - 10 °C. Chloroform (216.5 mL) was then added. More calcium carbonate was added until the pH was approximately 4.0. The chloroform was decanted off. The aqueous phase was washed twice with chloroform and the organic layers were combined. Morpholine (72.2 mL, 0.828 mol) was added to the chloroform solution until the pH was approximately 7.0. The reaction was left to stir for 1 hour at -5 °C and then was stirred at room temperature for 1 hour. The crude product was then triturated with ether yielding pure 24.4a in 59% yield.
Preparation of 4-(2-Chloro-5-methyl-1-oxy-pyridine -4-sulfonyl)-morpholine (24.5a)

[0337] Under nitrogen 3-methyl-4-(morpholinosulfonyl)pyridine 1-oxide (24.4a, 5.0 g, 0.01936 mol) was dissolved in phosphoryl chloride (15.00 mL). The reaction was heated to at
90 °C and left to stir for 1 hour. The reaction was cooled to room temperature and sat. sodium bicarbonate solution was added very slowly until the solution was basic. The mixture was extracted with DCM (2 x 200 mL), the combined organic layers dried over magnesium sulfate, and the solvent evaporated, yielding 24.5a in 44% yield.
Preparation of 2,2-DimethyI-N- [5-methyl-4-(morpholine-4-sulfonyl)-pyridin-2-yl] - propionamide (24A)

[0338] Compound 24A was prepared in 89% yield from 24.5a and 2,2-dimethylpropanamide following the procedure described for the synthesis of 18.4 in example 18 A.
1H NMR (CDCl3), δ 0.92 (t, J= 7 Hz, 3H), 1.29 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 2.56 (s, 3H), 3.31 (t, J= 5 Hz, 4H), 3.78 (t, J= 5 Hz, 4H), 8.03 (br s, 1H), 8.26 (s, 1H), 8.59 (s, 1H).
[0339] Example 24B was prepared from 24.5a and 2-cyclopropyl-2-methylpropanamide following the procedure described for the synthesis of 18.4 in example 18 A.
1H NMR (CDCl3), δ 0.51 (m, 2H), 0.65 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 2.56 (s, 3H), 3.31 (t, J= 5 Hz, 4H), 3.78 (t, J= 5 Hz, 4H), 8.27 (s, 1H), 8.59 (s, 1H), 8.64 (br s, 1H).
[0340] Examples 24C - 24F were prepared from 2,3-dimethyl-4-nitropyridine 1 -oxide (24.1b) and 4-nitro-6,7-dihydro-5H-cyclopenta[b]pyridine 1-oxide (24.1c) respectively following the procedures described in example 24A.
24C: 1H NMR (CDCl3), δ 0.91 (t, J= 7 Hz, 3H), 1.29 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 2.51 (s, 3H), 2.52 (s, 3H), 3.34 (t,J= 5 Hz, 4H), 3.78 (t, J= 5 Hz, 4H), 7.94 (br s, 1H), 8.45 (s, 1H).
24D: 1H NMR (CDCl3), δ 0.51 (m, 2H), 0.65 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 2.51 (s, 3H), 2.52 (s, 3H), 3.34 (t, J= 5 Hz, 4H), 3.79 (t, J= 5 Hz, 4H), 8.44 (s, 1H), 8.53 (br s, 1H).
24E: 1H NMR (CDCl3), δ 0.92 (t, J= 8 Hz, 3H), 1.29 (s, 6H), 1.67 (q, J= 8 Hz, 2H), 2.16 (m, 2H), 3.00 (t, J= 8 Hz, 2H), 3.19 (t, J= 5 Hz, 4H), 3.22 (t, J= 8 Hz, 2H), 3.76 (t, J= 5 Hz, 4H), 8.00 (br s, 1H), 8.34 (s, 1H).
24F: 1HNMR (CDCl3), δ 0.50 (m, 2H), 0.65 (m, 2H), 1.09 (m, 1H), 1.13 (s, 6H), 2.16 (m, 2H), 3.00 (t, J= 8 Hz, 2H), 3.19 (t, J= 5 Hz, 4H), 3.23 (t, J= 8 Hz, 2H), 3.76 (t, J= 5 Hz, 4H), 8.33 (s, 1H), 8.58 (br s, 1H).
EXAMPLE 24G
Preparation of N-cyclohexyl-5-methyl-4-(morpholinosulfonyl)pyridin-2-amine (24G)

[0341] To a solution of 4-(6-chloro-2,3-dimethyl-pyridine-4-sulfonyl)-morpholine (24.5b, 0.40 g, 0.00138 mol) in toluene (2 mL) was added cyclohexanamine (0.1888 mL, 0.001651 mol), sodium tert-butoxide (198.3 mg, 0.002064 mol), 2-di-t-butylphosphino-2'-methylbiphenyl (34.4 mg, 0.000110 mol) and palladium acetate (12.4 mg, 0.0000550 mol). The reaction was stirred at room temperature overnight, then heated at 100 °C for another 24 hours. The crude reaction mixture was directly purified by silica gel column chromatography using ethyl acetate-hexane (1:3) as eluent, yielding 180 mg (38%) of the pure coupling product 24G.
1H NMR (CDCl3), δ 1.24 (m, 3H), 1.40 (m, 2H), 1.65 (m, 1H), 1.75 (m, 2H), 2.02 (m, 2H), 2.37 (s, 3H), 2.42 (s, 3H), 3.19 (t, J= 5 Hz, 4H), 3.47 (m, 1H), 3.73 (t, J= 5 Hz, 4H), 4.55 (d, J= 8 Hz, 1H), 6.68 (s, 1H).
EXAMPLE 25A
Preparation of7V-(5-bromo-4-((methylamino)methyl)pyridin-2-yl)-2,2-dimethyl- butanamide (25.1)

[0342] To a 250 mL flask containing methylamine (9.88 g, 0.318 mol; as 30 mL of a 33 wt %, ~ 10.6 M solution in EtOH) at ~ 0 °C on an ice/water bath was added a clear solution of N-(5- bromo-4-bromomethylpyridin-2-yl)-2,2-dimethylbutanamide (2.3a2, 5.00 g, 0.0137 mol) in THF (10 mL, anhydrous), and the reaction was allowed to warm to room temperature with stirring overnight. The crude reaction mixture was concentrated and subsequently partitioned between EtOAc and saturated aq. NaHCO3. The layers were separated, and the aqueous phase was extracted once more with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated to 4.76 g of a clear, medium-brown oil.
The crude product was dissolved in a minimal amount of dichloromethane and purified with via flash chromatography using a 0-100% EtOAc/hexanes gradient yielding 2.3 g product 25.1 (53%). MS analysis m/z = 314+1.
[0343] Compounds 25A and 25 B were part of a parallel synthesis library and they were prepared from amine 25.1 following this general procedure using 4-fluorobenzene-1-sulfonyl chloride and 4-(trifluoromethyl)benzene-1-sulfonyl chloride respectively:
25A, B

[0344] Solutions of amine 25.1 (10 μmol/200 μL) and sulfonyl chlorides (15 μmol/100 μL) in NMP were prepared. Into a 96 well plate 200 μl of amine solution, 100 μL of sulfonyl chloride solution and 3 eq. of polystyrene bound DMAP were combined and shaken over night at room temperature. PL-trisamine resin was added (3 eq.) and the plate was shaken overnight. The reaction mixtures were filtered, the solvent evaporated and the plate reconstituted in DCM. PS- TsCl resin was added, the plate was shaken overnight then filtered and the residues screened without further purification. MS analysis m/z results see table 1.
EXAMPLE 25C
Preparation of N-(4-((benzyl(methyl)amino)methyl)-5-bromopyridin-2-yl)-2,2-dimethyl- butanamide (25.2)

[0345] Compound 25.1 was prepared from 2.3a2 and methyl benzyl amine in 35% yield following general method C. MS analysis m/z = 404+1.
Preparation ofN-(4-((benzyl(methyl)amino)methyl)-5-methyIpyridin-2-yl)-2-cyclo-propyl- 2-methylpropanamide (25C)

[0346] Compound 25C was prepared from 25.2 according to general procedure D, amide cleavage with potassium hydroxide in EtOH/water followed by reaction with acid chloride a6 according to general method A.
1H NMR (CDCl3), δ 0.49 (m, 2H), 0.63 (m, 2H), 1.10 (m, 1H), 1.13 (s, 6H), 2.16 (s, 3H), 2.27 (s, 3H), 3.47 (s, 2H), 3.53 (s, 2H), 7.30 (m, 5H), 8.01 (s, 1H), 8.24 (s, 1H), 8.46 (br s, 1H)
EXAMPLE 25D
Preparation of 2-cyclopropyl-2-methyl-N-(5-methyl-4-((methylamino)methyl)-pyridin-2- yl)propanamide (25.3)

[0347] Into a flask was added N-(4-((benzyl(methyl)ammo)methyl)-5-methylpyridin-2-yl)-2- cyclopropyl-2-methylpropanamide (25C, 1.36 g, 3.88 mmol) and ethanol (20 mL). Nitrogen was bubbled into the solution and Pd on carbon (10%, 130 mg) was added. The flask was evacuated, placed under H2 and stirred overnight at room temperature. A second portion of Pd on carbon (10%, 130 mg) was addded and stirring under an atmosphere of hydrogen was continued for 5 hours. The reaction mixture was filtered through celite, the solvent evaporated and the residue dried in vacuo overnight at room temperature yielding 25.3 quantitatively. MS analysis m/z = 261+1.
Preparation of 2-cyclopropyl-N-(4-((4-fluoro-N-methylphenylsulfonamido)methyI)-5- methylpyridin-2-yl)-2-methylpropanamide (25D)
 25D
25D
[0348] Into a round-bottom flask was added 2-cyclopropyl-2-methyl-N-(5-methyl-4-((methyl- amino)methyl)-pyridin-2-yl)propanamide (25.3, 45.0 mg, 0.172 mmol), N,N-diisopro- pylethylamine (45.0 uL, 0.258 mmol), and DCM (2 mL). The mixture was cooled to 0 °C and a solution of 4-fluorobenzenesulfonyl chloride (40.2 mg, 0.207 mmol) in DCM (1 mL) was added dropwise. The mixture was allowed to warm up to room temperature and stirred overnight. Then saturated NH4Cl solution (3mL) was added and the layers were separated. The aqueous layer was extracted once with DCM. The combined organic layers were washed with saturatedd NaHCO3 solution, dried overNa2SO4, filtered and the solvent evaporated yielding 88 mg pale yellow gum. The crude product was chromatographed on silica using a hexane/EtOAc gradient yielding 61 mg white solid 25D (85%).
MS analysis m/z = 419+1. 1H NMR (CDCl3), δ 0.48 (m, 2H), 0.62 (m, 2H), 1.06 (m, 1H), 1.10 (s, 6H), 2.38 (s, 3H), 2.58 (s, 3H), 4.12 (s, 2H), 7.26 (m, 2H), 7.86 (m, 2H), 8.00 (s, 1H), 8.09 (s, 1H), 8.48 (br s, 1H).
[0349] Example 25E was prepared from 25.3 and 4-(trifluoromethyl)benzene-1-sulfonyl chloride in 73% yield following the procedure described for 25D.
MS analysis m/z = 469+1. 1H NMR (CDCl3), δ 0.47 (m, 2H), 0.62 (m, 2H), 1.05 (m, 1H), 1.09 (s, 6H), 2.37 (s, 3H), 2.62 (s, 3H), 4.16 (s, 2H), 7.86 (d, J= 8 Hz, 2H), 7.98 (d, J= 8 Hz, 2H), 8.00 (s, 1H), 8.09 (s, 1H), 8.48 (br s, 1H).
EXAMPLE 25F
Preparation ofN-(5-bromo-4-(piperazin-1-ylmethyl)pyridin-2-yl)-2,2-dimethyl-butanami(le
(25.5)

[0350] Compound 25.5 was prepared from 25.4 (obtained from 2.3a2 and tert-butyl piperazine- 1-carboxylate and general method C) according to general method D, followed by deprotection with IM HCl. MS analysis m/z = 304+1.
Preparation of N-(2,3-dihydrobenzo [b] [1 ,4] dioxin-6-yl)-4-((2-(2,2-dimethyl-butanamido)-5- methylpyridin-4-yl)methyl)piperazine-1-carboxamide (25F)
25F

[0351] To amine 25.5 (3.04 mg, 0.01 mmol) dissolved in anhydrous THF (100 uL) was added 6-isocyanato-2,3-dihydrobenzo[b][1,4]dioxine (0.588 mg, 0.0140 mmol) dissolved in anhydrous THF (120 uL). The mixture was shaken overnight. Scavenger resin was added (3 eq. PS- trisamine), shaken for 3 hours, filtered and the solvent evaporated. MS analysis m/z = 481+1.
EXAMPLE 25G
Preparation of7V-(4-((4-benzylpiperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2-cyclopropyl-
2-methylpropanamide

[0352] Compound 25.7 was prepared according to general method D from 25.6 (obtained from 2.3a2, 1-benzylpiperazine and general method C), followed by amide cleavage with 6M HCl as described in example 3A and amide formation with acid chloride a6 according to general method A. MS analysis m/z = 406+1.
Preparation of7V-(4-((4-(1,2,3-thiadiazoIe-4-carbonyl)piperazin-1-yl)methyI)-5-methyl- pyridin-2-yl)-2-cyclopropyl-2-methylpropanamide (25G)

MS analysis m/z = 428+1. 1H NMR (CDCl3), δ 0.49 (m, 2H), 0.63 (m, 2H), 1.10 (m, 1H), 1.13 (s, 6H), 2.32 (s, 3H), 2.62 (m, 4H), 3.53 (s, 2H), 3.97 (m, 4H), 8.04 (s, 1H), 8.29 (s, 1H), 9.13 (s, 1H).
BIOLOGICAL ASSAYS:
In vitro Methods
[0354] Kj values in receptor binding experiments were determined by Cheng-Prusoff correction of IC50 values derived from automated nonlinear regression analysis of sigmoidal titration curves using a three-parameter modification (slope set to 1.0) of the four-parameter equation described in Cheng, Y.-C. and W. H. Prusoff, Biochem. Pharmacol. 22:3099-3108 (1973) and DeLean et al., Am. J. Physiol. 235:E97-E102 (1978).
[0355] EC50 values in functional assays were also derived from automated nonlinear regression analysis of sigmoidal titration curves using the three-parameter modification of the four- parameter equation.
Preparation of membranes for hCBl and hCB2 receptor binding and receptor-mediated stimulation of [35SIGTPYS binding
[0356] Chinese hamster ovary cells (CHO-Kl ), stably transfected with either hCB 1 or hCB2, were washed two times with cold PBS, scraped from 500 cm2 tissue culture plates, and pelleted by centrifugation at 1000 x g for 10 min. The supernatant was discarded and the pellet was resuspended in Tris assay buffer (50 mM Tris HCl, pH 7.8, containing 1.0 mM EGTA, 5.0 mM MgCl2, 10 mg/mL leupeptin, 10 mg/mL pepstatin A, 200 mg/mL bacitracin, and 0.5 mg/mL aprotinin), homogenized with a Polytron homogenizer (Brinkmann) at a setting of 1 for 20 sec and centrifuged at 38,000 x g for 20 min at 4°C. The pellet was resuspended in Tris assay buffer and aliquots of 1 mg protein/mL were stored at -80°C for further use.
Preparation of rat cerebellar membranes for cannabinoid receptor-mediated stimulation of T35SIGTPYS binding
[0357] Rat cerebella were excised and placed into homogenization buffer (50 mM Tris HCl, pH 7.4, containing 3 mM MgCl2 and 1 mM EGTA) and homogenized for 20 sec using a Polytron homogenizer at a setting of 1 and centrifuged at 4°C for 10 min at 48,000 x g. The supernatant was removed and the pellet was resuspended in homogenization buffer and centrifuged at 4°C for 10 min at 48,000 x g. The supernatant was removed and the pellets were resuspended in 50 mM Tris HCl, pH 7.4, containing 3 mM MgCl2 and 0.2 mM EGTA and stored as aliquots of 1 mg protein/mL at -80°C for further use.
Inhibition of CB receptor binding by test compounds
[0358] Binding assays were performed by incubating 0.2-0.6 nM (34,000-100,000 dpm) [3H]CP55940 with membranes prepared from cells expressing cloned human CB1 or CB2 receptors in buffer A (50 mM Tris HCl, pH 7.0, 5.0 mM MgCl2, 1.0 mM EGTA and 1.0 mg/mL fatty acid free bovine serum albumin). After incubation for 60 min at room temperature for the hCB2 binding assay or 120 min at 30°C for the hCB1 assay, the assays were filtered through GF/C filters that had been pre-soaked overnight in 0.5% (w/v) PEI and 0.1% BSA in water. The filters were rinsed 6 times with one mL of cold wash buffer (50 mM Tris HCl, pH 7.0, 5.0 mM MgCl2, 1.0 mM EGTA and 0.75 mg/mL fatty acid free bovine serum albumin), 30 μL of MicroScint 20 was added to each filter and the radioactivity on the filters determined by scintillation spectroscopy. Nonspecific binding was determined in the presence of 10 μM WIN55212-2.
Cannabinoid receptor-mediated stimulation of [35S]GTPyS binding
[0359] hCB1-Mediated stimulation of [35S]GTPyS binding was measured in a mixture containing 100-150 pM [35S]GTPyS, 150 mM NaCl, 45 mM MgCl2, 3 mM GDP, 0.4 mM DTT, 1 mM EGTA, 1 mg/mL fatty acid free BSA, 25 μg of membrane protein and agonist in a total volume of 250 μL of buffer A in 96 well Basic Flashplates (Perkin Elmer). After incubation at room temperature for 2 hours the plates were centrifuged at 800 X g at 4°C for 5 min and the radioactivity bound to the membranes was determined by scintillation spectrometry using the Topcount (Perkin Elmer).
[0360] hCB2-Mediated [35S]GTPyS binding was measured in the same way except the assay mixture contained 10 mM GDP and the incubation time was 6 hours. [35S]GTPyS binding in rat cerebellar homogenate was determined in a mixture containing 40-60 pM [35S]GTPyS,
homogenate assay buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA), 100 mM NaCl, 10 mM MgCl2, 100 mM GDP, 20 μg homogenate protein/well and agonist in a total volume of 250 μL in 96 well Basic Flashplates (Perkin Elmer). After incubation at 30°C for 2 hours, the plates were centrifuged at 800 X g at 4°C for 5 min and the radioactivity bound to the membrane was determined by scintillation spectrometry using the Topcount (Perkin Elmer).
In vitro Results
[0361] Compounds IA-I ID, listed in Table 1, were tested for their affinity toward the human cloned CB1 and CB2 receptors. AU ligands tested bound to the human CB1 and/or CB2 receptor with affinity ranging from 0.1 - 10000 nM. These ligands displayed various degrees of selectivity, CB2 vs. CB1. The functional potency of selected ligands was also evaluated in vitro. Some compounds were found to exhibit agonist activity at CB1 and/or CB2 receptors. For example, compound 2B (K1 (CB1) = 3820 nM, K1 (CB2) = 18.6 nM) was found to possess no in vitro CB1 receptor agonist activity but good in vitro CB2 receptor agonist potency (EC50 = 33 nM). Some compounds were also found to exhibit antagonist activity towards the CB2 receptor. Compound 2C (K1 (CB1) > 1000 nM, K1 (CB2) = 33 nM) was found to possess in vitro CB2 receptor antagonist activity (-2% stimulation in the [35S]GTPyS assay).
Table 1. List of compounds

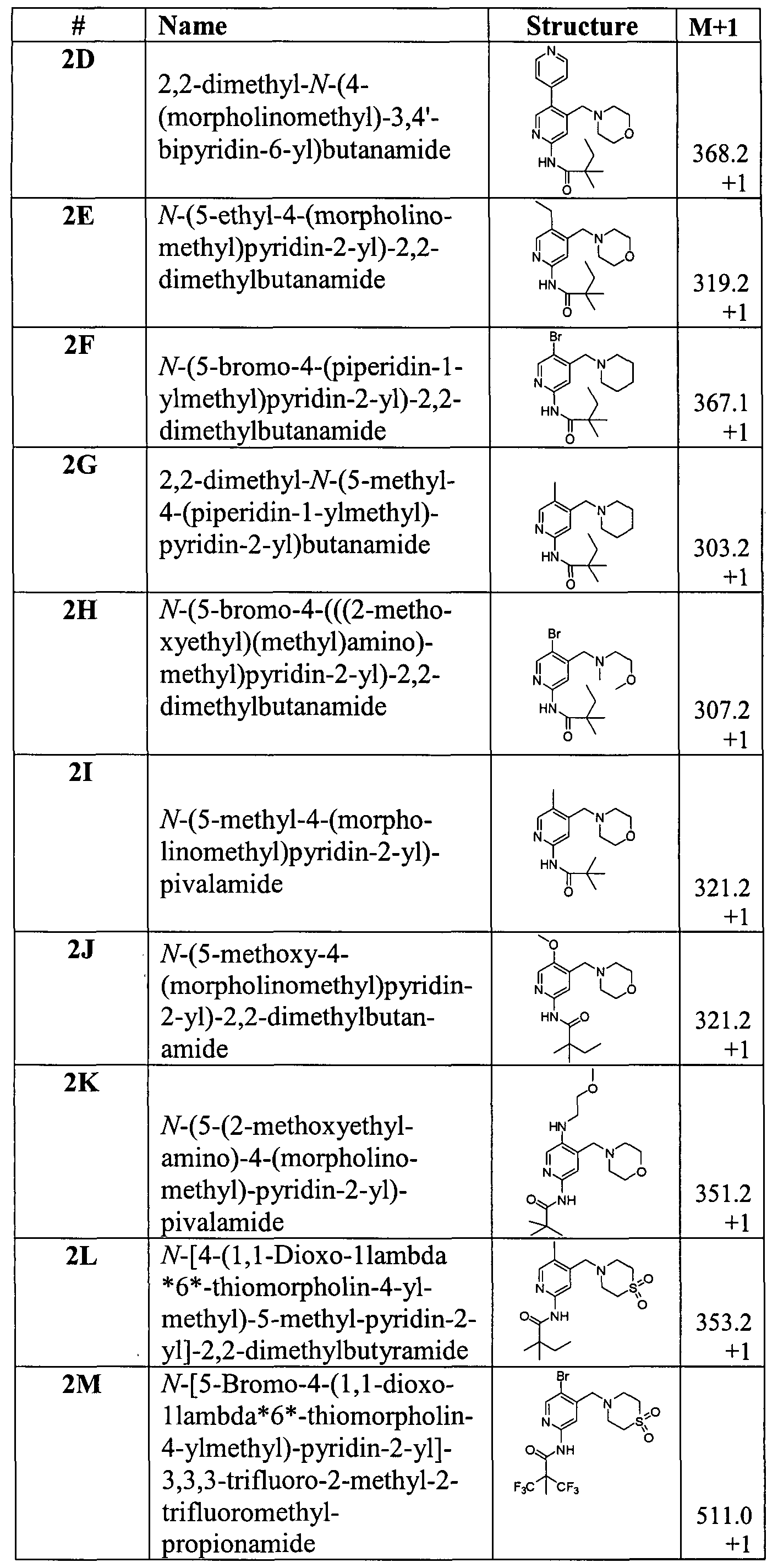













[0362] When ranges are used herein for physical properties, such as molecular weight, or chemical properties, such as chemical formulae, all combinations and subcombinations of ranges and specific embodiments therein are intended to be included.
[0363] The disclosures of each patent, patent application and publication cited or described in this document are hereby incorporated herein by reference, in their entirety.
[0364] Those skilled in the art will appreciate that numerous changes and modifications can be made to the preferred embodiments of the invention and that such changes and modifications can be made without departing from the spirit of the invention. It is, therefore, intended that the appended claims cover all such equivalent variations as fall within the true spirit and scope of the invention.
Claims
1. A compound according to formula I:

A is a ring atom or a bond;
D is cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, or -NR2R3; E is N or CR12; Y is N or CR6;
R is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -ORX, -SRX, -NRyRz, F, Cl, or Br, or R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R8)(R9)]n-D; provided that: when R1 or D independently includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7-membered ring having 0 or 1 heteroatom ring members, and when R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic ring, then the formed monocyclic ring has 0 or 1 heteroatom ring members; Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl; or Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
R2 and R3 are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl, or R2 and R3 when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
Z is -C(=O)-, -N(R5)-, -C(=O)N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-;
R4 is:

R5 and R5a are each independently H or alkyl;
Rc is H, alkyl, or aryl;
Rd and Re are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
R6 and R12 are each independently H, F, Cl, or alkyl, or R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring substituted with -[C(R8)(R9)]n-D;
R is H, F, or alkyl, or R and R taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
R8 and R9 are each independently H or alkyl; each R9a is independently H, alkyl, aryl, -C(=O)-R11, -C(=O)-OR11, -[C(R11)(R11^s-C(=O)-OR11, -SO2R", or -C(=O)N(R1 ^R11; each R10 is independently H, alkyl, or aryl; each R11 is independently H or alkyl; n and r are each independently 0, 1, 2, or 3; and s is 1, 2, 3, or 4; provided that:
(1) only one of E and Y is N;
(2) at least one of R1, R6, and R7 is other than F;
(3) when R7 is F, then at least one of R1 and R6 is other than F, Cl, or Br;
(4) when R1 is -ORX, then E is N;
(5) when n is 0, then -A-[C(R8)R9)]n-D is other than aryl or heteroaryl;
(6) at least one of R2 and R3 is other than H;
(7) at least two of Rc, Rd, and Re are other than H; and
(8) no more than one pair of R7 and R12, R1 and R7 , R1 and A, and R6 and A form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
2. A compound according to claim 1, wherein A is a bond.
3. A compound according to claim 2, wherein D is -NR2R3.
4. A compound according to claim 3, wherein R2 and R3 are each independently alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl, or R2 and R3 when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, Or -C(=O)-N(R10)-.
5. A compound according to claim 4, wherein R2 and R3 are each independently alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl.
6. A compound according to claim 5, wherein at least one of R and R is aralkyl.
7. A compound according to claim 6, wherein at least one of R2 and R3 is benzyl.
8. A compound according to claim 4, wherein R2 and R3 when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-.
9. A compound according to claim 8, wherein R and R , taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein one of the heterocycloalkyl ring carbon atoms is optionally replaced by -O-, -S-, or -N(R9a)-.
10. A compound according to claim 9, wherein R2 and R3, taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein one of the heterocycloalkyl ring carbon atoms is replaced by -O-, -S-, or -N(R9a)-.
11. A compound according to claim 1 , wherein E is N or CH.
12. A compound according to claim 11 , wherein E is N.
13. A compound according to claim 1, wherein Y is N or CH.
14. A compound according to claim 13, wherein Y is CH.
15. A compound according to claim 1, wherein R7 is H or alkyl.
16. A compound according to claim 15, wherein R7 is H or methyl optionally substituted with one or more fluorine atoms.
17. A compound according to claim 16, wherein R7 is H, CH3, or CF3.
18. A compound according to claim 1, wherein R1 is H, alkyl, cycloalkyl, aryl, heteroaryl, -ORX, or Br.
19. A compound according to claim 18, wherein R1 is H, C1-C3alkyl, cycloalkyl, aryl, heteroaryl, -ORX, or Br.
20. A compound according to claim 1, wherein R1 and R7 taken together with the atoms through which they are attached form monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring.
21. A compound according to claim 20, wherein R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl ring.
22. A compound according to claim 1, wherein R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, substituted with -[C(R )(R )]n-D-
23. A compound according to claim 22, wherein R1 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl ring substituted with -[C(R8)(R9)]n-D.
24. A compound according to claim 1, wherein R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring.
25. A compound according to claim 24, wherein R7 and R12 taken together with the atoms through which they are attached form a monocyclic 4- to 8-membered cycloalkyl ring.
26. A compound according to claim 1 , wherein R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, substituted with -[C(R8)(R9)]n-D.
27. A compound according to claim 26, wherein R6 and the ring atom A taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl ring substituted with -[C(R8)(R9)]n-D.
28. A compound according to claim 1 , wherein R5 is H or C1-C3alkyl.
29. A compound according to claim 1, wherein Z is -C(=O)N(R5)- or -N(R5)C(=O)-.
30. A compound according to claim 29, wherein Z is -N(R5)C(=O)-.
31. A compound according to claim 29, wherein R5 is H or C1.C3alkyl.
32. A compound according to claim 31 , wherein R5 is H.
33. A compound according to claim 1 , wherein each Ra, Rb, Rd, and Re is independently H or alkyl.
34. A compound according to claim 33, wherein Ra and Rb are each H.
35. A compound according to claim 33, wherein Rd and Re are each independently alkyl.
36. A compound according to claim 35, wherein Rd and Re are each C1-C6alkyl.
37. A compound according to claim 36, wherein Rc is C1-C6alkyl.
38. A compound according to claim 37, wherein at least two of Rc, Rd, and Re are each independently C1-Caalkyl.
39. A compound according to claim 38, wherein Rc, Rd, and Re are each independently C1-C3alkyl.
40. A compound according to claim 37, wherein at least one of Rc, Rd, and Re is C1-C6alkyl substituted with at least one fluoro.
41. A compound according to claim 33, wherein R4 is:

42. A compound according to claim 1 , wherein Rd and Re taken together with the carbon atom to which they are attached form a carbocyclic ring.
43. A compound according to claim 42, wherein Rd and Re taken together with the carbon atom to which they are attached form a monocyclic carbocyclic ring.
44. A compound according to claim 43, wherein Rd and Re taken together with the carbon atom to which they are attached form a monocyclic C3-C8carbocyclic ring.
45. A compound according to claim 44, wherein Rd and Re taken together with the carbon atom to which they are attached form a monocyclic C3-C6carbocyclic ring.
46. A compound according to claim 45, wherein Rd and Re taken together with the carbon atom to which they are attached form a substituted monocyclic C3-C6carbocyclic ring.
47. A compound according to claim 46, wherein Rd and Re taken together with the carbon atom to which they are attached form a monocyclic C3-C6carbocyclic ring substituted with at least one C1-C6alkyl.
48. A compound according to claim 47, wherein Rd and Re taken together with the carbon atom to which they are attached form a monocyclic C3-Cόcarbocyclic ring substituted with at least one C1-C3alkyl.
49. A compound according to claim 43, wherein R4 is:
wh r 2; and r 3. 
50. A compound according to claim 42, wherein Rd and Re, taken together with the carbon atom to which they are attached, form a 3- to 12-membered carbocyclic ring, wherein the carbocyclic ring is substituted with 0-5 groups each independently selected from C1.C4alkyl and C1.C4alkoxyl.
51. A compound according to claim 50, wherein the carbocyclic ring is bicycloalkyl or tricycloalkyl.
52. A compound according to claim 51, wherein the carbocyclic ring is bicycloalkyl.
53. A compound according to claim 52, wherein the bicycloalkyl ring is substituted with 1-3 C1.C4alkyl groups.
54. A compound according to claim 1, wherein Rc is H, C!.C3alkyl, or C6aryl.
55. A compound according to claim 1, wherein r is 0, 1, or 2.
56. A compound according to claim 55, wherein r is 0 or 1.
57. A compound according to claim 56, wherein r is 0.
58. A compound according to claim 1 , of formula II:
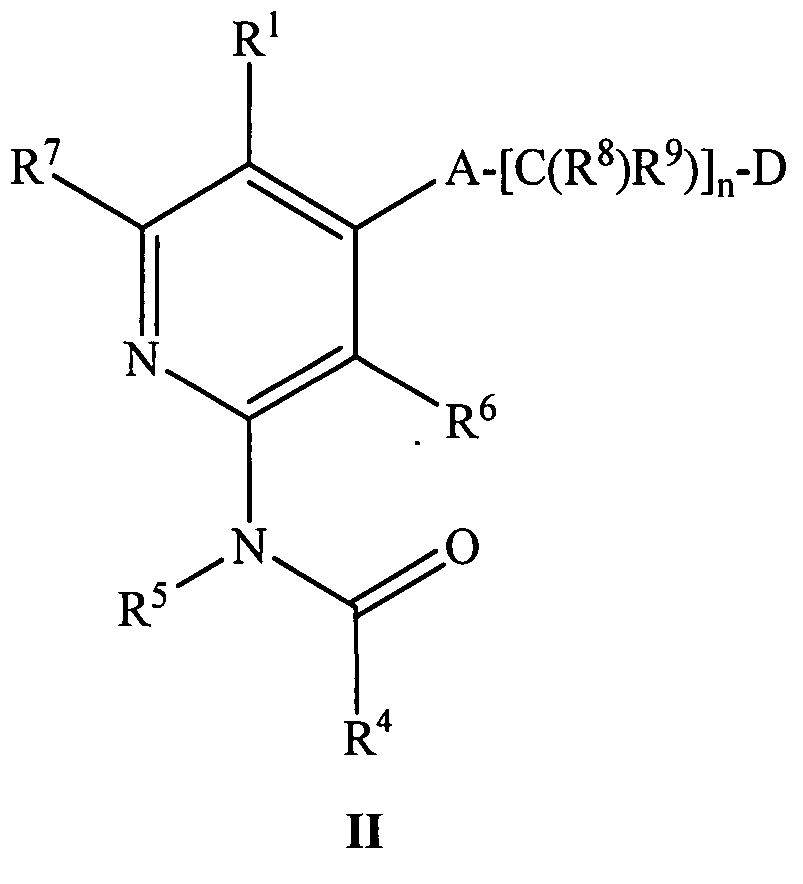
59. A compound according to claim 58, wherein A is a bond.
60. A compound according to claim 59, wherein n is 0 or 1 and D is -NR2R3.
61. A compound according to claim 60, wherein n is 1.
62. A compound according to claim 60, wherein R2 and R3, taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein one of the heterocycloalkyl ring carbon atoms is optionally replaced by -O-, -S-, or - N(R9a)-.
63. A compound according to claim 62, wherein R5 and R6 are each H.
64. A compound according to claim 63, wherein Rc, Rd, and Re are each C1-C3alkyl.
65. A compound according to claim 63, wherein R7 is H or alkyl and R1 is alkyl, alkoxy, or Br.
66. A compound according to claim 64, wherein R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl ring.
67. A compound according to claim 1 , of formula III:

68. A compound according to claim 67, wherein A is a bond.
69. A compound according to claim 68, wherein n is 0 or 1 and D is -NR 2r R>3
70. A compound according to claim 69, wherein n is 1.
71. A compound according to claim 69, wherein R2 and R3, taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein one of the heterocycloalkyl ring carbon atoms is optionally replaced by -O-, -S-, or -N(R9a)-.
72. A compound according to claim 71, wherein Rc, Rd, and Re are each C].C3alkyl.
73. A compound according to claim 72, wherein R7 is H or alkyl and R1 is alkyl, alkoxy, or Br.
74. A compound according to claim 72, wherein R1 and R7 taken together with the atoms through which they are attached form a monocyclic 6-membered aryl ring.
75. A compound according to claim 1, wherein R1 is -ORX.
76. A compound according to claim 75, wherein R1 is alkoxy.
77. A compound according to claim 76, wherein R1 is Q-Qalkoxy.
78. A compound according to claim 77, wherein R1 is C1-C3alkoxy.
79. A compound according to claim 78, wherein R1 is methoxy.
80. A compound of claim 1, selected from the group consisting of: 2-methyl-N-(4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide; N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide; N-(5-isopropyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)-5-phenyl-pyridin-2-yl)-butanamide; N-(5-cyclohexyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(5-(furan-3-yl)-4-(morpholinomethyl)-pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(5-methyl-4-(piperidin-1-ylmethyl)-pyridin-2-yl)-butanamide; N-(4-((-N'-(2-methoxyethyl)-N'-(methyl)amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide; 1,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-cyclohexanecarboxamide; 2,2,3,3-tetramethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
1,2,2,3,3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(6-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-morpholinoquinolin-2-yl)-butanamide;
2-methyl-ΛL(4-morpholinoquinolin-2-yl)-cyclohexanecarboxamide;
2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-6-(trifluoromethyl)pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(5-morpholino-5,6,7,8-tetrahydroisoquinolin-3-yl)-butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
81. A compound according to claim 80, selected from the group consisting of: 2-methyl-N-(4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide; N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide; N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(6-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide; Λ/-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(4-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide; 2,2-dimethyl-N-(4-morpholinoquinolin-2-yl)-butanamide; 2-methyl-N-(4-morpholinoquinolin-2-yl)-cyclohexanecarboxamide; 2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
82. A compound according to claim 81, selected from the group consisting of: N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide; N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 7V-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 7V-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(4-morpholinoquinolin-2-yl)-butanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
83. A compound according to claim 82, selected from the group consisting of: 2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide; N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide; and
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;or a pharmaceutically acceptable salt thereof.
84. A pharmaceutical composition, comprising: a pharmaceutically acceptable carrier; and a compound of claim 1.
85. A pharmaceutical composition according to claim 84, wherein the compound of claim 1 is selected from the group consisting of:
2-methyl-N-(4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide;
7V-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-isopropyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5-phenyl-pyridin-2-yl)-butanamide;
N-(5-cyclohexyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-(furan-3-yl)-4-(morpholinomethyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(piperidin-1-ylmethyl)-pyridin-2-yl)-butanamide;
N-(4-((-N '-(2-methoxyethyl)-N '-(methyl)amino)methyl)-5 -methylpyridin-2-yl)-2,2- dimethylbutanamide ;
2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide;
1,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-cyclohexanecarboxamide;
2,2,3,3-tetramethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
1,2,2,3, 3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(6-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
7V-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-7V-(4-morpholinoquinolin-2-yl)-butanamide;
2-methyl-N-(4-morpholinoquinolin-2-yl)-cyclohexanecarboxamide;
2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-6-(trifluoromethyl)pyridin-2-yl)- butanamide; 2,2-dimethyl-N-(5-morpholino-5,6,7,8-tetrahydroisoquinolin-3-yl)-butanamide; 2,2-dimethyl-N-(4-(moipholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; 2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
86. A pharmaceutical composition according to claim 84, further comprising at least one cannabinoid.
87. A pharmaceutical composition according to claim 86, wherein the cannabinoid is Δ -tetrahydrocannabinol or cannabidiol.
88. A pharmaceutical composition according to claim 84, further comprising at least one opioid.
89. A pharmaceutical composition according to claim 88, wherein said opioid is selected from the group consisting of alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil and tramadol, and mixtures thereof.
90. A pharmaceutical composition according to claim 84, further comprising at least one analgesic.
91. A pharmaceutical composition according to claim 90, wherein the analgesic is a COX2 inhibitor, aspirin, acetaminophen, ibuprophen, or naproxen, or a mixture thereof.
92. A pharmaceutical composition according to claim 84, further comprising at least one agent selected from the group consisting of an anti-seizure agent, an anti-depressant, an NMDA receptor antagonist, an ion channel antagonist, a nicotinic receptor agonist, and an anti- Parkinson's agent.
93. A pharmaceutical composition according to claim 92 wherein said anti-seizure agent is carbamazepine, gabapentin, lamotrigine, or phenytoin, or a mixture thereof.
94. A pharmaceutical composition according to claim 92 wherein said anti-depressant is amitryptiline.
95. A pharmaceutical composition according to claim 92, wherein said anti-Parkinson's agent is deprenyl, amantadine, levodopa, or carbidopa, or a mixture thereof.
96. A method of binding cannabinoid receptors in a patient in need thereof, comprising the step of: administering to said patient an effective amount of a compound of claim 1.
97. A method according to claim 96, wherein the compound of claim 1 is selected from the group consisting of:
2-methyl-N-(4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide;
N-(5-bromo-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-isopropyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5-phenyl-pyridin-2-yl)-butanamide;
N-(5-cyclohexyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-(furan-3-yl)-4-(morpholinomethyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(piperidin-1-ylmethyl)-pyridin-2-yl)-butanamide;
N-(4-((-N'-(2-methoxyethyl)-N'-(methyl)amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
2-methyl-7V-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)-cyclohexanecarboxamide;
1,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)-cyclohexanecarboxamide;
2,2,3,3-tetramethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
1,2,2,3,3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)- cyclopropanecarboxamide;
N-(5-bromo-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5,6-dimethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-Λ'-(6-methyl-4-(morpholinomethyl)-pyridin-2-yl)-butanamide;
N-(5-bromo-4-methyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; N-(4,5-dimethyl-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-6-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-N-(4-methyl-6-(morpholinomethyl)-pyridin-2-yl)-butanamide;
2,2-dimethyl-ΛL(4-morpholinoquinolin-2-yl)-butanamide;
2-methyl-N-(4-morpholinoquinolin-2-yl)-cyclohexanecarboxamide;
2,2-dimethyl-N-(4-(morpholinomethyl)quinolin-2-yl)-butanamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-6-(trifluoromethyl)pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(5-morpholino-5,6,7,8-tetrahydroisoquinolin-3-yl)-butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide;
2,2-dimethyl-N-(4-(morpholinomethyl)-5,6,7,8-tetrahydroquinolin-2-yl)-butanamide; and
7V-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
98. A method according to claim 96, wherein the cannabinoid receptors are selected from the group consisting of CB1 and CB2 cannabinoid receptors.
99. A method according to claim 96, wherein the cannabinoid receptors are located in the central nervous system.
100. A method according to claim 96, wherein the cannabinoid receptors are located peripherally to the central nervous system.
101. A method according to claim 100, wherein the compound selectively binds CB2 cannabinoid receptors relative to CB1 receptors.
102. A method according to claim 96, wherein the binding agonizes the activity of the cannabinoid receptors.
103. A method according to claim 96, wherein the binding antagonizes the activity of the cannabinoid receptors.
104. A method according to claim 96, wherein the binding inversely agonizes the activity of the cannabinoid receptors.
105. A method according to claim 96, further comprising administering to said patient an effective amount of at least one cannabinoid.
106. A method according to claim 105, wherein said cannabinoid is Δ9-tetrahydrocannabinol or cannabidiol.
107. A method according to claim 96, further comprising administering to said patient an effective amount of at least one opioid.
108. A method according to claim 107, wherein said opioid is selected from alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil and tramadol, and a mixture thereof.
109. A method according to claim 96 which is for the treatment of a disease or disorder selected from the group consisting of a gastrointestinal disorder, inflammation, an auto-immune disease, an immune-related disorder, pain, hypertension, a neurodegenerative disease, a neurological disorder, and a combination thereof.
110. A method according to claim 98 which is for providing cardioprotection against ischemic or reperfusion effects, inhibiting mechanical hyperalgesia associated with nerve injury, inducing apoptosis in malignant cells, modulating appetite, or a combination thereof.
111. A method according to claim 109 which is for the treatment of pain.
112. A method according to claim 111, further comprising administering to said patient at least one cannabinoid.
113. A method according to claim 111, wherein said pain is inflammatory pain, neuropathic pain, visceral pain, surgical pain, post-surgical pain, cancer related pain, or a combination thereof.
114. A method according to claim 113, further comprising administering to said patient codeine, carbamazepine, gabapentin, lamotrigine, phenytoin, amitryptiline, an NMDA receptor antagonist, an ion channel antagonist, a nicotinic receptor agonist, or a mixture thereof.
115. A method according to claim 109, which is for the treatment of a gastrointestinal disorder.
116. A method according to claim 115, wherein the gastrointestinal disorder is nausea, vomiting, loss of appetite, cachexia, diarrhea, inflammatory bowel disease, irritable bowel syndrome, or a combination thereof.
117. A method according to claim 109, which is for the treatment of an auto-immune disease.
118. A method according to claim 117, wherein the auto-immune disease is multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, systemic lupus erythematosus, myasthenia gravis, diabetes mellitus type I, osteoporosis, or a combination thereof.
119. A method according to claim 109, which is for the treatment of a neurological disorder.
120. A method according to claim 119, wherein said neurological disorder is stroke, migraine, cluster headache, or a combination thereof.
121. A method according to claim 109, which is for the treatment of an immune-related disorder.
122. A method according to claim 121, wherein said immune-related disorder is asthma, chronic pulmonary obstructive disorder, emphysema, bronchitis, allergy, tissue rejection in organ transplants, celiac disease, Sjogren's syndrome, or a combination thereof.
123. A method according to claim 109, which is for the treatment of a neurodegenerative disease.
124. A method according to claim 123, wherein said neurodegenerative disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis, or a combination thereof.
125. A method according to claim 123, further comprising administering to said patient deprenyl, amantadine, levodopa, or carbidopa.
126. A method according to claim 110, which is for providing cardioprotection against ischemic or reperfusion effects.
127. A method according to claim 126, wherein the ischemic or reperfusion effect is arrhythmia or hypertension.
128. A method according to claim 110, which is for inducing apoptosis in malignant cells.
129. A method according to claim 128, wherein the apoptosis occurs in vitro.
130. A method according to claim 128, wherein the apoptosis occurs in vivo.
131. A compound according to formula Ia:

wherein:
A is -S(=O)2- or a bond;
D is -N(H)-S(=O)2-aryl, -N(Rv)-S(=O)2-aryl, -N(H)aralkyl, -N(CH3)aralkyl, or heterocycloalkyl, in which the heterocycloalkyl ring contains at least one nitrogen atom or dioxo-thio group; provided that: when A is -S(=O)2-, then n is 0 and D is -N(H)aralkyl, -N(CH3)aralkyl, or heterocycloalkyl, wherein the heterocycloalkyl group in D contains at least one nitrogen atom, and A is attached to D through the heterocycloalkyl nitrogen ring atom; and when A is a bond, then D is -N(H)-S(=O)2-aryl, -N(Rv)-S(=O)2-aryl, or a heterocycloalkyl ring containing at least one dioxo-thio group;
E is N or CR12;
Y is N or CR6;
R1 is H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroaralkyl, -ORX, -SRX, -NRyRz, F, Cl, or Br; provided that when R1 includes a cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring moiety, then the included ring moiety is a monocyclic 3- to 7- membered ring having 0 or 1 heteroatom ring members,
Rv is C1-3unsubstituted alkyl;
Rx, Ry, and Rz are each independently H, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, or heteroaralkyl; or Ry and Rz, when taken together with the nitrogen atom to which they are attached, form a monocyclic 3- to 7-membered heterocycloalkyl ring in which 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, -N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-;
Z is -N(R5)-, -N(R5)C(=O)-, or -N(R5)C(=O)N(R5a)-;
R4 is:

R5 and R5a are each independently H or alkyl;
Rc is H, alkyl, or aryl;
Rd and Re are each independently H or alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring;
R6 and R12 are each independently H, F, Cl, or alkyl;
R7 is H, F, or alkyl, or R1 and R7 taken together with the atoms through which they are connected form a monocyclic 4- to 7-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring, or R7 and R12 taken together with the atoms through which they are connected form a monocyclic 4- to 8-membered cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring;
R and R are each independently H or alkyl; each R9a is independently H, alkyl, aryl, -C(=O)-R11, -C(=O)-OR11, -[C(R11)(R11^s-C(=O)-OR11, -SO2R11, Or -C(=O)N(R1^R11; each R10 is independently H, alkyl, or aryl; each R11 is independently H or alkyl; n and r are each independently 0, 1, 2, or 3; and s is 1, 2, 3, or 4; provided that:
(1) only one of E and Y is N;
(2) at least two of Rc, Rd, and Re are other than H; and
(3) no more than one pair of R7 and R12, and R1 and R7 form a monocyclic cycloalkyl, aryl, heterocycloalkyl, or heteroaryl ring; or a pharmaceutically acceptable salt thereof.
132. A compound according to claim 131, wherein A is a bond.
133. A compound according to claim 131, wherein A is -S(=O)2-.
134. A compound according to claim 132, wherein D is -N(H)-S(=O)2-aryl or -N(RV)-S(=O)2- aryl.
135. A compound according to claim 132, wherein D is heterocycloalkyl containing at least one nitrogen atom or dioxo-thio group.
136. A compound according to claim 135, wherein D is a heterocycloalkyl ring containing at least one nitrogen atom.
137. A compound according to claim 136, wherein D is morpholinyl, piperidinyl, or 1,1- dioxo-thiomorpholinyl.
138. A compound according to claim 134, wherein D is -N(H)-S(=O)2-aryl or -N(CH3)- S(=O)2-aryl.
139. A compound according to claim 133, wherein D is -N(H)-aralkyl or -N(CH3)-aralkyl.
140. A compound according to claim 139, wherein D is -N(H)-benzyl or -N(CH3)-benzyl.
141. A compound according to claim 132, wherein Z is -N(R5)-, or-N(R5)C(=O)-.
142. A compound according to claim 132, wherein r is 0.
143. A compound according to claim 132, wherein n is 0 or 1.
144. A compound according to claim 132, wherein E is N and Y is CH.
145. A compound according to claim 144, wherein Rc, Rd, and Re are each independently aallkkyyll;; oorr RRdd aanndd F Re taken together with the carbon atom to which they are attached form a carbocyclic ring.
146. A compound according to claim 145, wherein n is 1, A is a bond, D is 1,1- dioxothiomorpholinyl and Z is -N(H)C(=O)-.
147. A compound according to claim 145, wherein n and r are each 0, Z is -N(H)C(=O)- or -NH-, A is -S(=O)2-, and D is optionally substituted morpholinyl.
148. A compound according to claim 145, wherein A is a bond, n is 1, Z is -N(H)C(=O)-, and D is -N(H)-S(=O)2-aryl, -N(CH3)-S(=O)2-aryl.
149. A compound according to claim 148, wherein the aryl moiety of D is phenyl, optionally substituted with at least one halo or haloalkyl group.
150. A compound according to claim 1, wherein Z is -N(R5)-.
151. A compound according to claim 150, wherein A is a bond, n is 1 , R and R are each H and D is NR2R3, wherein R2 and R3 taken together with the nitrogen atom to which they are attached form a monocyclic 3- to 7-membered heterocycloalkyl ring, wherein 1 or 2 of the heterocycloalkyl ring carbon atoms independently may each be optionally replaced by -O-, -S-, - N(R9a)-, -N(R10)-C(=O)-, or -C(=O)-N(R10)-.
152. A compound according to claim 151, wherein D is morpholinyl.
153. A compound according to claim 151, wherein R5 is H and R4 is:  wherein
wherein
Rc, Rd, and Re are each independently alkyl, or taken together with the carbon atom to which they are attached form a carbocyclic ring.
154. A compound according to claim 60, wherein R1 is bromo and R2 and R3 are each independently alkyl.
155. A compound according to claim 61, wherein Rc, Rd, and Re are each independently alkyl.
156. A compound according to claim 155, wherein R2 is aralkyl or cycloalkyl.
157. A compound according to claim 156, wherein R2 is aralkyl.
158. A compound according to claim 157, wherein R3 is H or alkyl.
159. A compound according to claim 63, wherein n is 0 or 1.
160. A compound according to claim 159, wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a monocyclic 5- to 7-membered heterocycloalkyl ring, wherein 1 of the heterocycloalkyl ring carbon atoms is optionally replaced by -O-, -S-, or -N(R9a)-.
161. A compound according to claim 160, wherein Rd and Re are taken together with the carbon atom to which they are attached to form a carbocyclic ring.
162. A compound according to claim 160, wherein Rc, Rd, and Re are each independently alkyl.
163. A compound according to claim 160, wherein D is morpholinyl.
164. A compound according to claim 161, wherein D is morpholinyl and n is 1.
165. A compound according to claim 148, wherein R1 and R7 are taken together with the atoms through which they are connected to form a monocyclic 4- to 7-membered cycloalkyl ring.
166. A compound according to claim 64, wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a monocyclic 5- to 7-membered heterocycloalkyl ring, wherein 1 of the heterocycloalkyl ring carbon atoms is optionally replaced by -O-, -S-, or -N(R9a)-.
167. A compound according to claim 166, wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a monocyclic 5- to 7-membered heterocycloalkyl ring, wherein 1 of the heterocycloalkyl ring carbon atoms is replaced by -O-, - S-, or -N(R9a)-.
168. A compound according to claim 166, wherein the monocyclic 5- to 7-membered heterocycloalkyl ring is fused to a benzene ring.
169. A compound according to claim 167, wherein one of the heterocycloalkyl ring carbon atoms is replaced by -O-.
170. A compound according to claim 169, wherein R1 is alkyl.
171. A compound according to claim 65, wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a monocyclic 5- to 7-membered heterocycloalkyl ring, wherein 1 of the heterocycloalkyl ring carbon atoms is optionally replaced by -O-, -S-, or -N(R9a)-.
172. A compound according to claim 171, wherein R2 and R3 are taken together with the nitrogen atom to which they are attached to form a monocyclic 5- to 7-membered heterocycloalkyl ring, wherein 1 of the heterocycloalkyl ring carbon atoms is replaced by -O-, - S-, or -N(R9a)-.
173. A compound according to claim 172, wherein one of the heterocycloalkyl ring carbon atoms is replaced by -O-.
174. A compound according to claim 173, wherein Rc, Rd, and Re are each independently alkyl.
175. A compound according to claim 174, wherein R1 is alkyl or alkoxy.
176. A compound according to claim 162, wherein the monocyclic 3- to 7-membered heterocycloalkyl ring is substituted with at least one hydroxy, alkyl, dialkylamino, halo, heteroarylcarbonyl, or alkylcarbonyl group.
177. A compound according to claim 176, wherein R1 is alkyl or alkoxy.
178. A compound according to claim 72, wherein R1 is H, Br, or alkyl, and R7 is H or alkyl.
179. A compound according to claim 1, selected from the group consisting of: N-(5-(2-methoxyethyl-amino)-4-(morpholinomethyl)-pyridin-2-yl)-pivalamide; N-(5-bromo-4-(thiomorpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 2,2-dimethyl-N-(5-methyl-4-(thiomorpholinomethyl)-pyridin-2-yl)butanamide; N-(4-((1,4-oxazepan-4-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide; N-(5-amino-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 7V-(5-bromo-4-((4-methylpiperazin-1-yl)methyl)-pyridin-2-yl)-2,2-dimethylbutanamide; N-(4-((4-acetylpiperazin-1-yl)methyl)-5-bromopyridin-2-yl)-2,2-dimethylbutanamide; N-(5-bromo-4-((4-phenylpiperazin-1-yl)methyl)-pyridin-2-yl)-2,2-dimethylbutanamide; 2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide; 1,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)cyclohexanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide; 1,2,2,3,3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclo- propanecarboxamide;
2,2-diethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclopropanecarboxamide;
3,3,3-trifluoro-2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)-2- (trifluoromethyl)propanamide;
2-cyclopropyl-Λf-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)propanamide;
2-cyclopropyl-2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)propanamide;
2-ethyl-2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclopentanecarboxamide;
2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)pentanamide;
4,4,4-trifluoro-2,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide; 1-ethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclobutanecarboxamide; 1-ethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclopentanecarboxamide; 1-ethyl-7V-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclohexanecarboxamide;
2,2-diethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)butanamide;
2,2,3,3-tetramethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]-pyridin-2- y^cyclopropanecarboxamide;
2-cyclopropyl-2-methyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]- pyridin-2-yl)propanamide;
N-(4-((1,4-oxazepan-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-2,2- dimethylbutanamide ;
N-(4-((l ,4-oxazepan-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)- 1 - ethylcyclopentanecarboxamide;
N-(4-((1,4-oxazepan-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-2,2- diethylbutanamide;
N-cycloheptyl-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-amine;
7V-( 1 -methoxypropan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta- [b]pyridin- 2-amine;
N-(bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]- pyridin-2-amine;
N-(5-isopropyl-4-(morpholinomethyl)-pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2,2, 3,3- tetramethylcyclopropanecarboxamide;
N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)- 1,2,2, 3,3- pentamethylcyclopropanecarboxamide;
2-cyclopropyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2-methylpropanamide;
2-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)-2-methylbutanamide; 2,2-diethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)butanamide;
N-(5-ethyl-4-(morpholino-methyl)pyridin-2-yl)-3,3,3-trifluoro-2-methyl-2- (trifluoromethyl)propanamide; 1-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-7V-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)cyclopentanecarboxamide; 1-ethyl-N-(5-ethyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2,3,3- tetramethylcyclopropanecarboxamide;
2-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2-methylbutanamide;
2,2-diethyl-N-(5-methoxy-4-(morpholinomethyl)-pyridin-2-yl)butanamide;
3,3,3-trifluoro-7V-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)-2-methyl-2- (trifluoromethyl)-propanamide; 1-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)cyclopentanecarboxamide; 1-ethyl-N-(5-methoxy-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2, 3,3- tetramethylcyclopropanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-l ,2,2, 3,3- pentamethylcyclopropanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2-ethyl-2-methylbutanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-diethylbutanamide;
Ν-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-3,3,3-trifluoro-2-methyl-2- (trifluoromethyl)propanamide;
2-cyclopropyl-N-(5-ethoxy-4-(morpholinomethyl)-pyridin-2-yl)propanamide;
2-cyclopropyl-yV-(5-ethoxy-4-(morpholinomethyl)-pyridin-2-yl)-2-methylpropanamide;
N-(5 -ethoxy-4-(morpholinomethyl)pyridin-2-yl)- 1 -ethylcyclobutanecarboxamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-1-ethylcyclopentanecarbox-amide;
7V-(5 -ethoxy-4-(morpholinomethyl)pyridin-2-yl)- 1 -ethylcyclohexanecarboxamide;
N-(5-(methylthio)-4-(morpholinomethyl)pyridin-2-yl)pivalamide; 1-ethyl-N-(5-(methylthio)-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide;
N-(5-(ethylthio)-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-methoxy-6-methyl-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide; 3,3,3-trifluoro-2-methyl-N-(4-(morpholinomethyl)-5-propoxypyridin-2-yl)-2- (trifluoromethyl)propanamide;
N-(4-(isoindolin-2-yl-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((3,4-dihydroiso-quinolin-2(1H)-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((benzyl(ethyl)amino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((benzyl(methyl)-amino)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((benzyl(isopropyl)-amino)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(pyrrolidin-1-ylmethyl)-pyridin-2-yl)butanamide;
N-(4-((3-hydroxypyrrolidin-1-yl)methyl)-5-methyl-pyridin-2-yl)-2,2-dimethyl- butanamide;
N-(4-((3-(dimethylamino)-pyrrolidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((3-fluoropyrrolidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-(thiazolidin-3-ylmethyl)-pyridin-2-yl)butanamide;
N-(4-((2,5-dimethyl-2,5-dihydro-1H-pyrrol-1-yl)-methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
2,2-dimethyl-N-(5 -methyl-4-((4-methylpiperidin- 1 -yl)methyl)pyridin-2-yl)-butanamide;
N-(4-((3,5-dimethylpiperidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide ;
N-(4-((4-hydroxypiperidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((3-fluoropiperidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-((4-methylpiperazin-1-yl)methyl)pyridin-2-yl)-butanamide;
N-(4-((4-ethylpiperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((4-acetylpiperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
7V-(4-((bis(2-methoxyethyl)-amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-(((2-(dimethylamino)-ethyl)(ethyl)amino)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide;
N-(4-((sec-butylamino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((cyclopentylamino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((cyclohexylamino)-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-((2-methylcyclohexylamino)methyl)pyridin-2- yl)butanamide; 2,2-dimethyl-N-(5-methyl-4-((1-phenylethylamino)methyl)pyridin-2-yl)-butanamide;
2,2-dimethyl-ΛL(5-methyl-4-(1-morpholinopropyl)-pyridin-2-yl)butanamide;
N-(4-((benzyl(methyl)-amino)methyl)-5-methylpyridin-2-yl)-2-cyclopropyl-2- methylpropanamide;
N-(2,3-dihydrobenzo-[b][1,4]dioxin-6-yl)-4-((2-(2,2-dimethylbutanamido)-5- methylpyridin-4-yl)-methyl)piperazine- 1 -carboxamide;
N-(4-((4-(1,2,3-thiadiazole-4-carbonyl)piperazin-1-yl)methyl)-5-methylpyridin-2-yl)-2- cyclopropyl-2-methylpropanamide;
N-(4-(4-fluorophenethyl)-5,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5',6l-dimethyl-2,4'-bipyridin-2'-yl)-2,2-dimethylbutanamide;
N-(4-benzyl-5,6-dimethylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-bromo-4-phenylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(5-methoxy-4-phenylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((1-acetylpiperidin-4-yl)methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide;
N-(4-((1-acetylpiperidin-4-yl)methyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2-yl)-2,2- dimethylbutanamide;
2,2-dimethyl-N-(5-methyl-4-((tetrahydro-2H-pyran-4-yl)methyl)pyridin-2-yl)- butanamide;
2,2-dimethyl-N-(7-(4-(morpholinomethyl)-2,3-dihydrofuro-[3,2-b]pyridin-5- yl)butanamide; and
2,2-dimethyl-N-(5-methyl-4-(2-morpholinoethyl)-pyridin-2-yl)butanamide; or a pharmaceutically acceptable salt thereof.
180. A compound according to claim 179, selected from the group consisting of: N-((li?,2i?,45)-bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H- cyclopenta[b]-pyridin-2-amine;
(S)-N-(4-((3-fluoropyrrolidin-1-yl)methyl)-5-methylpyridin-2-yl)-2,2- dimethylbutanamide; and
(S)-2,2-dimethyl-7V-(5-methyl-4-((1-phenylethylamino)methyl)pyridin-2-yl)-butanamide; or a pharmaceutically acceptable salt thereof.
181. A compound according to claim 179, selected from the group consisting of: 1,2-dimethyl-N-(5-methyl-4-(morpholinomethyl)-pyridin-2-yl)cyclohexanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclobutanecarboxamide; 1-ethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)cyclohexanecarboxamide; 1,2,2,3,3-pentamethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2- yl)cyclopropanecarboxamide;
2,2-diethyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)butanamide;
2-cyclopropyl-2-methyl-N-(5-methyl-4-(morpholinomethyl)pyridin-2-yl)propanamide; 1-ethyl-N-(4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-2- yl)cyclohexanecarboxamide;
N-(bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H-cyclopenta[b]- pyridin-2-amine;
N-((li?,2i?,41S)-bicyclo-[2.2.1]heptan-2-yl)-4-(morpholinomethyl)-6,7-dihydro-5H- cyclopenta[b]-pyridin-2-amine;
N-(5-ethyl-4-(morpholino-methyl)pyridin-2-yl)-2,2-dimethylbutanamide;
3 ,3 ,3 -trifluoro-N-(5 -methoxy-4-(morpholinomethyl)pyridin-2-yl)-2-methyl-2- (trifluoromethyl)-propanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-2,2-dimethylbutanamide;
Ν-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)-3,3,3-trifluoro-2-methyl-2- (trifluoromethyl)propanamide;
N-(5-ethoxy-4-(morpholinomethyl)pyridin-2-yl)- 1 -ethylcyclopentanecarboxamide; and
N-(4-(isoindolin-2-yl-methyl)-5-methylpyridin-2-yl)-2,2-dimethylbutanamide; or a pharmaceutically acceptable salt thereof.
182. A compound of claim 131, selected from the group consisting of:
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-yl-methyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyr amide;
N-[5-Bromo-4-(1,1-dioxo-1-lambda*6*-thiomorpholin-4-ylmethyl)-pyridin-2-yl]-3,3,3- trifluoro-2-methyl-2-trifluoromethylpropionamide;
N-[4-(l , 1-Dioxo- 1 -lambda *6*-thiomorpholin-4-ylmethyl)-5-methylpyridin-2-yl]-3,3, 3- trifluoro-2-methyl-2-trifluoromethylpropionamide;
2-Cyclopropyl-N-[4-(1,1-dioxo-1-lambda*6*-thiomorpholin-4-ylmethyl)-5- methylpyridin-2-yl]-isobutyramide;
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-yl-methyl)-5-methoxypyridin -2-yl]-2,2- dimethylbutyr amide;
N-(5-bromo-4-((4-fluoro-N-methylphenylsulfonamido)methyl)pyridin-2-yl)-2,2- dimethylbutanamide; N-(5-bromo-4-((N-methyl-4-(trifluoromethyl)phenylsulfonamido)methyl)pyridin-2-yl)- 2,2-dimethylbutanamide;
N-(4-((3-chloro-N-methylphenylsulfonamido)methyl)-5-methylpyridin-2-yl)-2- cyclopropyl-2-methylpropanamide;
2-cyclopropyl-N-(4-((4-fluoro-N-methylphenylsulfonamido)methyl)-5-methylpyridin-2- yl)-2-methylpropanamide;
2-cyclopropyl-2-methyl-N-(5-methyl-4-((N-methyl-4- (trifluoromethyl)phenylsulfonamido)methyl)pyridin-2-yl)propanamide;
N-[4-(1,1-Dioxo-1-lambda *6*-thiomoφholin-4-ylmethyl)-5,6-dimethylpyridin-2-yl]- 2,2-dimethylbutyramide;
2-Cyclopropyl-N-[4-(l , 1 -dioxo- 1 -lambda*6*-thiomoφholin-4-ylmethyl)-6,7-dihydro- 5H--[[ 1 yrindin-2-yl]-isobutyramide;
N-[4-( 1 , 1 -Dioxo-hexahydro- 1 -thiopyran-4-ylmethyl)-5 -methylpyridin-2-yl] -2,2- dimethylbutyramide;
2,2-dimethyl-N-(5-methyl-4-(moφholinosulfonyl)-pyridin-2-yl)butanamide; 2-cyclopropyl-2-methyl-N-(5-methyl-4-(moφholinosulfonyl)pyridin-2-yl)-propanamide;
N-(5,6-dimethyl-4-(moφholinosulfonyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2-cyclopropyl-N-(5,6-dimethyl-4-(moφholinosulfonyl)pyridin-2-yl)-2- methylpropanamide;
2,2-dimethyl-7V-(4-(moφholinosulfonyl)-6,7-dihydro-5H-cyclopenta-[b]pyridin-2- yl)butanamide;
2-cyclopropyl-2-methyl-N-(4-(moφholinosulfonyl)-6,7-dihydro-5H-cyclo- penta[b]pyridin-2-yl)propanamide; and
N-cyclohexyl-5,6-dimethyl-4-(moφholinosulfonyl)-pyridin-2-amine; or a pharmaceutically acceptable salt thereof.
183. A compound of claim 178, selected from the group consisting of: N-[4-(1,1-Dioxo-1-lambda *6*-thiomoφholin-4-yl-methyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyramide; and
N-[4-(l , 1-Dioxo- 1 -lambda *6*-thiomoφholin-4-ylmethyl)-5,6-dimethylpyridin-2-yl]- 2 ,2-dimethylbutyramide ; or a pharmaceutically acceptable salt thereof.
184. A pharmaceutical composition, comprising: a pharmaceutically acceptable carrier; and a compound of claim 131.
185. A pharmaceutical composition, comprising: a pharmaceutically acceptable carrier; and a compound of claim 182.
186. A pharmaceutical composition according to claim 184, further comprising at least one cannabinoid.
187. A pharmaceutical composition according to claim 186, wherein the cannabinoid is Δ9-tetrahydrocannabinol or cannabidiol.
188. A pharmaceutical composition according to claim 184, further comprising at least one opioid.
189. A pharmaceutical composition according to claim 188, wherein said opioid is selected from the group consisting of alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil and tramadol, and mixtures thereof.
190. A pharmaceutical composition according to claim 184, further comprising at least one analgesic.
191. A pharmaceutical composition according to claim 190, wherein the analgesic is a COX2 inhibitor, aspirin, acetaminophen, ibuprophen, or naproxen, or a mixture thereof.
192. A pharmaceutical composition according to claim 184, further comprising at least one agent selected from the group consisting of an anti-seizure agent, an anti-depressant, an NMDA receptor antagonist, an ion channel antagonist, a nicotinic receptor agonist, and an anti- Parkinson's agent.
193. A pharmaceutical composition according to claim 192, wherein said anti-seizure agent is carbamazepine, gabapentin, lamotrigine, or phenytoin, or a mixture thereof.
194. A pharmaceutical composition according to claim 192, wherein said anti-depressant is amitryptiline.
195. A pharmaceutical composition according to claim 192, wherein said anti-Parkinson's agent is deprenyl, amantadine, levodopa, or carbidopa, or a mixture thereof.
196. A method of binding cannabinoid receptors in a patient in need thereof, comprising the step of: administering to said patient an effective amount of a compound of claim 131.
197. A method according to claim 196, wherein the compound administered is selected from the group consisting of:
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-yl-methyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyramide;
N-[5-Bromo-4-(1,1-dioxo-1-lambda*6*-thiomorpholin-4-ylmethyl)-pyridin-2-yl]-3,3,3- trifluoro-2-methyl-2-trifluoromethylpropionamide;
N- [4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-ylmethyl)-5 -methylpyridin-2-yl]-3 ,3 ,3 - trifluoro-2-methyl-2-trifluoromethylpropionamide;
2-Cyclopropyl-N-[4-(1,1-dioxo-1-lambda*6*-thiomorpholin-4-ylmethyl)-5- methylpyridin-2-yl] -isobut yramide ;
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-yl-methyl)-5-methoxypyridin -2-yl]-2,2- dimethylbutyramide;
N-(5-bromo-4-((4-fluoro-N-methylphenylsulfonamido)methyl)pyridin-2-yl)-2,2- dimethylbutanamide;
N-(5-bromo-4-((N-methyl-4-(trifluoromethyl)phenylsulfonamido)methyl)pyridin-2-yl)- 2,2-dimethylbutanamide;
N-(4-((3-chloro-N-methylphenylsulfonamido)methyl)-5-methylpyridin-2-yl)-2- cyclopropyl-2-methylpropanamide;
2-cyclopropyl-N-(4-((4-fluoro-N-methylphenylsulfonamido)methyl)-5-methylpyridin-2- yl)-2-methylpropanamide;
2-cyclopropyl-2-methyl-7V-(5-methyl-4-((N-methyl-4- (trifluoromethyl)phenylsulfonamido)methyl)pyridin-2-yl)propanamide;
N-[4-(1,1-Dioxo-1-lambda *6*-thiomorpholin-4-ylmethyl)-5,6-dimethylpyridin-2-yl]- 2,2-dimethylbutyr amide;
2-Cyclopropyl-N-[4-(l , 1 -dioxo- 1 -lambda*6*-thiomorpholin-4-ylmethyl)-6,7-dihydro- 5H-[ 1]pyrindin-2-yl]-isobutyramide;
7V-[4-(l , 1 -Dioxo-hexahydro-1 -thiopyran-4-ylmethyl)-5-methylpyridin-2-yl]-2,2- dimethylbutyramide; 2,2-dimethyl-N-(5-methyl-4-(morpholinosulfonyl)-pyridin-2-yl)butanamide; 2-cyclopropyl-2-methyl-N-(5-methyl-4-(morpholinosulfonyl)pyridin-2-yl)-propanamide;
N-(5,6-dimethyl-4-(morpholinosulfonyl)pyridin-2-yl)-2,2-dimethylbutanamide;
2-cyclopropyl-N-(5,6-dimethyl-4-(morpholinosulfonyl)pyridin-2-yl)-2- methylpropanamide;
2,2-dimethyl-N-(4-(morpholinosulfonyl)-6,7-dihydro-5H-cyclopenta-[b]pyridin-2- yl)butanamide;
2-cyclopropyl-2-methyl-N-(4-(morpholinosulfonyl)-6,7-dihydro-5H-cyclo- penta[b]pyridin-2-yl)propanamide; and
N-cyclohexyl-5,6-dimethyl-4-(morpholinosulfonyl)-pyridin-2-amine; or a pharmaceutically acceptable salt thereof.
198. A method according to claim 196, wherein the cannabinoid receptors are selected from the group consisting of CB1 and CB2 cannabinoid receptors.
199. A method according to claim 196, wherein the cannabinoid receptors are located in the central nervous system.
200. A method according to claim 196, wherein the cannabinoid receptors are located peripherally to the central nervous system.
201. A method according to claim 200, wherein the compound selectively binds CB2 cannabinoid receptors relative to CB1 receptors.
202. A method according to claim 296, wherein the binding agonizes the activity of the cannabinoid receptors.
203. A method according to claim 196, wherein the binding antagonizes the activity of the cannabinoid receptors.
204. A method according to claim 196, wherein the binding inversely agonizes the activity of the cannabinoid receptors.
205. A method according to claim 196, further comprising administering to said patient an effective amount of at least one cannabinoid.
206. A method according to claim 205, wherein said cannabinoid is Δ9-tetrahydrocannabinol or cannabidiol.
207. A method according to claim 196, further comprising administering to said patient an effective amount of at least one opioid.
208. A method according to claim 207, wherein said opioid is selected from alfentanil, buprenorphine, butorphanol, codeine, dezocine, dihydrocodeine, fentanyl, hydrocodone, hydromorphone, levorphanol, loperamide, meperidine (pethidine), methadone, morphine, nalbuphine, oxycodone, oxymorphone, pentazocine, propiram, propoxyphene, sufentanil and tramadol, and a mixture thereof.
209. A method according to claim 196 which is for the treatment of a disease or disorder selected from the group consisting of a gastrointestinal disorder, inflammation, an auto-immune disease, an immune-related disorder, pain, hypertension, a neurodegenerative disease, a neurological disorder, and a combination thereof.
210. A method according to claim 196 which is for providing cardioprotection against ischemic or reperfusion effects, inhibiting mechanical hyperalgesia associated with nerve injury, inducing apoptosis in malignant cells, modulating appetite, or a combination thereof.
211. A method according to claim 209 which is for the treatment of pain.
212. A method according to claim 211, further comprising administering to said patient at least one cannabinoid.
213. A method according to claim 211, wherein said pain is inflammatory pain, neuropathic pain, visceral pain, surgical pain, post-surgical pain, cancer related pain, or a combination thereof.
214. A method according to claim 213, further comprising administering to said patient codeine, carbamazepine, gabapentin, lamotrigine, phenytoin, amitryptiline, an NMDA receptor antagonist, an ion channel antagonist, a nicotinic receptor agonist, or a mixture thereof.
215. A method according to claim 209, which is for the treatment of a gastrointestinal disorder.
216. A method according to claim 215, wherein the gastrointestinal disorder is nausea, vomiting, loss of appetite, cachexia, diarrhea, inflammatory bowel disease, irritable bowel syndrome, or a combination thereof.
217. A method according to claim 209, which is for the treatment of an auto-immune disease.
218. A method according to claim 217, wherein the auto-immune disease is multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, systemic lupus erythematosus, myasthenia gravis, diabetes mellitus type I, osteoporosis, or a combination thereof.
219. A method according to claim 209, which is for the treatment of a neurological disorder.
220. A method according to claim 219, wherein said neurological disorder is stroke, migraine, cluster headache, or a combination thereof.
221. A method according to claim 209, which is for the treatment of an immune-related disorder.
222. A method according to claim 221, wherein said immune-related disorder is asthma, chronic pulmonary obstructive disorder, emphysema, bronchitis, allergy, tissue rejection in organ transplants, celiac disease, Sjogren's syndrome, or a combination thereof.
223. A method according to claim 209, which is for the treatment of a neurodegenerative disease.
224. A method according to claim 223, wherein said neurodegenerative disease is Parkinson's disease, Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis, or a combination thereof.
225. A method according to claim 223, further comprising administering to said patient deprenyl, amantadine, levodopa, or carbidopa.
226. A method according to claim 210, which is for providing cardioprotection against ischemic or reperfusion effects.
227. A method according to claim 226, wherein the ischemic or reperfusion effect is arrhythmia or hypertension.
228. A method according to claim 210, which is for inducing apoptosis in malignant cells.
229. A method according to claim 228, wherein the apoptosis occurs in vitro.
230. A method according to claim 228, wherein the apoptosis occurs in vivo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US86652206P | 2006-11-20 | 2006-11-20 | |
| US60/866,522 | 2006-11-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008063625A2 true WO2008063625A2 (en) | 2008-05-29 |
| WO2008063625A3 WO2008063625A3 (en) | 2008-07-17 |
Family
ID=39315581
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/024220 WO2008063625A2 (en) | 2006-11-20 | 2007-11-19 | Pyridine compounds and methods of their use |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008063625A2 (en) |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100152254A1 (en) * | 2007-02-21 | 2010-06-17 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Alpha-halo- and alpha-alkyl-cyclopropylcarboxy compounds and uses thereof |
| EP2253364A2 (en) | 2009-05-18 | 2010-11-24 | Wacker Chemie AG | Silicone antifoam particles |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US8642604B2 (en) | 2006-04-04 | 2014-02-04 | The Regents Of The University Of California | Substituted pyrazolo[3,2-d]pyrimidines as anti-cancer agents |
| US8697709B2 (en) | 2008-10-16 | 2014-04-15 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8703777B2 (en) | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8785454B2 (en) | 2009-05-07 | 2014-07-22 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8809349B2 (en) | 2011-01-10 | 2014-08-19 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US20140296294A1 (en) * | 2011-12-15 | 2014-10-02 | University Of Pittsburgh -- Of The Commonwealth System Of Higher Education | Novel cannabinoid receptor 2 (cb2) inverse agonists and therapeutic potential for multiple myeloma and osteoporosis bone diseases |
| JP2014527075A (en) * | 2011-09-12 | 2014-10-09 | エフ.ホフマン−ラ ロシュ アーゲー | 5-Cycloalkyl- or 5-heterocyclyl-nicotinamides |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9056877B2 (en) | 2011-07-19 | 2015-06-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US9296742B2 (en) | 2008-09-26 | 2016-03-29 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US9321772B2 (en) | 2011-09-02 | 2016-04-26 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| JP2016539174A (en) * | 2013-12-02 | 2016-12-15 | シエナ・バイオテック・エス・ピー・エー | S1P3 antagonist |
| US9629843B2 (en) | 2008-07-08 | 2017-04-25 | The Regents Of The University Of California | MTOR modulators and uses thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10131668B2 (en) | 2012-09-26 | 2018-11-20 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pYRAZINES for modulation of IRE1 |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| CN110128422A (en) * | 2019-01-04 | 2019-08-16 | 金凯(辽宁)化工有限公司 | The synthetic method of 5- methoxyl group -7- azaindole |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| WO2021092262A1 (en) * | 2019-11-05 | 2021-05-14 | Dermira, Inc. | Mrgprx2 antagonists and uses thereof |
| US11110096B2 (en) | 2014-04-16 | 2021-09-07 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11147818B2 (en) | 2016-06-24 | 2021-10-19 | Infinity Pharmaceuticals, Inc. | Combination therapies |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4173565A (en) * | 1977-12-23 | 1979-11-06 | Toms River Chemical Corporation | Rubine disazo acid dyes for polyamides |
| US4499094A (en) * | 1982-04-27 | 1985-02-12 | Pharmuka Laboratoires | Derivatives of arene and hetero-arene carboxamides and their use as medicaments |
| WO2004029026A1 (en) * | 2002-09-27 | 2004-04-08 | Glaxo Group Limited | Pyridine derivatives as cb2 receptor modulators |
| WO2005080342A1 (en) * | 2004-02-24 | 2005-09-01 | Glaxo Group Limited | Pyridine derivatives and their use as cb2 receptor modulators |
| WO2006041900A2 (en) * | 2004-10-07 | 2006-04-20 | Cytovia, Inc. | SUBSTITUTED N-ARYL-1H-PYRAZOLO[3,4-b]QUINOLIN-4-AMINES AND ANALOGS AS ACTIVATORS OF CASPASES AND INDUCERS OF APOPTOSIS |
-
2007
- 2007-11-19 WO PCT/US2007/024220 patent/WO2008063625A2/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4173565A (en) * | 1977-12-23 | 1979-11-06 | Toms River Chemical Corporation | Rubine disazo acid dyes for polyamides |
| US4499094A (en) * | 1982-04-27 | 1985-02-12 | Pharmuka Laboratoires | Derivatives of arene and hetero-arene carboxamides and their use as medicaments |
| WO2004029026A1 (en) * | 2002-09-27 | 2004-04-08 | Glaxo Group Limited | Pyridine derivatives as cb2 receptor modulators |
| WO2005080342A1 (en) * | 2004-02-24 | 2005-09-01 | Glaxo Group Limited | Pyridine derivatives and their use as cb2 receptor modulators |
| WO2006041900A2 (en) * | 2004-10-07 | 2006-04-20 | Cytovia, Inc. | SUBSTITUTED N-ARYL-1H-PYRAZOLO[3,4-b]QUINOLIN-4-AMINES AND ANALOGS AS ACTIVATORS OF CASPASES AND INDUCERS OF APOPTOSIS |
Non-Patent Citations (3)
| Title |
|---|
| BARUFFI ET AL: "fitotossicita di n,n-di-sec.butilamidi acidibencoici monosostituiti" FARMACO ED. SCI., vol. 26, 1971, pages 891-896, XP009099767 * |
| BIEHL E R ET AL: "SYNTHESIS OF CERTAIN META DERIVATIVES OF N-ALKYLANILINES VIA ARYNE REACTIONS IN PRIMARY ALIPHATIC AMINE SOLVENTS" JOURNAL OF ORGANIC CHEMISTRY, AMERICAN CHEMICAL SOCIETY. EASTON, US, vol. 36, no. 21, 1 September 1971 (1971-09-01), page 3252/3253, XP000653762 ISSN: 0022-3263 * |
| C. BOLM ET AL: "enantioselective synthesis of optically active pyrimdine derivatives and c2-symmetric 2,2'-bipyridines" CHEMISCHE BERICHTE, vol. 125, 1992, pages 1169-1190, XP002479054 * |
Cited By (94)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| US9493467B2 (en) | 2006-04-04 | 2016-11-15 | The Regents Of The University Of California | PI3 kinase antagonists |
| US8642604B2 (en) | 2006-04-04 | 2014-02-04 | The Regents Of The University Of California | Substituted pyrazolo[3,2-d]pyrimidines as anti-cancer agents |
| US8518979B2 (en) * | 2007-02-21 | 2013-08-27 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Alpha-halo- and alpha-alkyl-cyclopropylcarboxy compounds and uses thereof |
| US20100152254A1 (en) * | 2007-02-21 | 2010-06-17 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Alpha-halo- and alpha-alkyl-cyclopropylcarboxy compounds and uses thereof |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US11433065B2 (en) | 2008-01-04 | 2022-09-06 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8785456B2 (en) | 2008-01-04 | 2014-07-22 | Intellikine Llc | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US9655892B2 (en) | 2008-01-04 | 2017-05-23 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9822131B2 (en) | 2008-01-04 | 2017-11-21 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9216982B2 (en) | 2008-01-04 | 2015-12-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8703777B2 (en) | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9637492B2 (en) | 2008-03-14 | 2017-05-02 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9828378B2 (en) | 2008-07-08 | 2017-11-28 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9629843B2 (en) | 2008-07-08 | 2017-04-25 | The Regents Of The University Of California | MTOR modulators and uses thereof |
| US9790228B2 (en) | 2008-09-26 | 2017-10-17 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US9296742B2 (en) | 2008-09-26 | 2016-03-29 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US8697709B2 (en) | 2008-10-16 | 2014-04-15 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| US8785454B2 (en) | 2009-05-07 | 2014-07-22 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US9315505B2 (en) | 2009-05-07 | 2016-04-19 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8461221B2 (en) | 2009-05-18 | 2013-06-11 | Wacker Chemie Ag | Silicone antifoam particles |
| EP2253364A2 (en) | 2009-05-18 | 2010-11-24 | Wacker Chemie AG | Silicone antifoam particles |
| DE102009003187A1 (en) | 2009-05-18 | 2010-11-25 | Wacker Chemie Ag | Silicone antifoam particles |
| US9522146B2 (en) | 2009-07-15 | 2016-12-20 | Intellikine Llc | Substituted Isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US9206182B2 (en) | 2009-07-15 | 2015-12-08 | Intellikine Llc | Substituted isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US8569323B2 (en) | 2009-07-15 | 2013-10-29 | Intellikine, Llc | Substituted isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US9181221B2 (en) | 2010-05-21 | 2015-11-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9738644B2 (en) | 2010-05-21 | 2017-08-22 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9388183B2 (en) | 2010-11-10 | 2016-07-12 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| USRE46621E1 (en) | 2011-01-10 | 2017-12-05 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9290497B2 (en) | 2011-01-10 | 2016-03-22 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9840505B2 (en) | 2011-01-10 | 2017-12-12 | Infinity Pharmaceuticals, Inc. | Solid forms of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1 (2H)-one and methods of use thereof |
| US10550122B2 (en) | 2011-01-10 | 2020-02-04 | Infinity Pharmaceuticals, Inc. | Solid forms of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one and methods of use thereof |
| US11312718B2 (en) | 2011-01-10 | 2022-04-26 | Infinity Pharmaceuticals, Inc. | Formulations of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one |
| US8809349B2 (en) | 2011-01-10 | 2014-08-19 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9605003B2 (en) | 2011-07-19 | 2017-03-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9056877B2 (en) | 2011-07-19 | 2015-06-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9718815B2 (en) | 2011-07-19 | 2017-08-01 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9115141B2 (en) | 2011-08-29 | 2015-08-25 | Infinity Pharmaceuticals, Inc. | Substituted isoquinolinones and methods of treatment thereof |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9546180B2 (en) | 2011-08-29 | 2017-01-17 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9321772B2 (en) | 2011-09-02 | 2016-04-26 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9895373B2 (en) | 2011-09-02 | 2018-02-20 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| JP2014527075A (en) * | 2011-09-12 | 2014-10-09 | エフ.ホフマン−ラ ロシュ アーゲー | 5-Cycloalkyl- or 5-heterocyclyl-nicotinamides |
| US20140296294A1 (en) * | 2011-12-15 | 2014-10-02 | University Of Pittsburgh -- Of The Commonwealth System Of Higher Education | Novel cannabinoid receptor 2 (cb2) inverse agonists and therapeutic potential for multiple myeloma and osteoporosis bone diseases |
| US9376380B2 (en) * | 2011-12-15 | 2016-06-28 | University of Pittsburgh—of the Commonwealth System of Higher Education | Cannabinoid receptor 2 (CB2) inverse agonists and therapeutic potential for multiple myeloma and osteoporosis bone diseases |
| US9255108B2 (en) | 2012-04-10 | 2016-02-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9527847B2 (en) | 2012-06-25 | 2016-12-27 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US10822340B2 (en) | 2012-09-26 | 2020-11-03 | The Regents Of The University Of California | Substituted imidazolopyrazine compounds and methods of using same |
| US10131668B2 (en) | 2012-09-26 | 2018-11-20 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pYRAZINES for modulation of IRE1 |
| US11613544B2 (en) | 2012-09-26 | 2023-03-28 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pyrazines for modulation of IRE1 |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US10639281B2 (en) | 2013-08-12 | 2020-05-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9828377B2 (en) | 2013-10-04 | 2017-11-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10329299B2 (en) | 2013-10-04 | 2019-06-25 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| JP2016539174A (en) * | 2013-12-02 | 2016-12-15 | シエナ・バイオテック・エス・ピー・エー | S1P3 antagonist |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10792254B2 (en) | 2013-12-17 | 2020-10-06 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10675286B2 (en) | 2014-03-19 | 2020-06-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11541059B2 (en) | 2014-03-19 | 2023-01-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11944631B2 (en) | 2014-04-16 | 2024-04-02 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11110096B2 (en) | 2014-04-16 | 2021-09-07 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US10941162B2 (en) | 2014-10-03 | 2021-03-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10253047B2 (en) | 2014-10-03 | 2019-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| US11247995B2 (en) | 2015-09-14 | 2022-02-15 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US11939333B2 (en) | 2015-09-14 | 2024-03-26 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11147818B2 (en) | 2016-06-24 | 2021-10-19 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| CN110128422B (en) * | 2019-01-04 | 2022-03-18 | 金凯(辽宁)生命科技股份有限公司 | Synthesis method of 5-methoxy-7-azaindole |
| CN110128422A (en) * | 2019-01-04 | 2019-08-16 | 金凯(辽宁)化工有限公司 | The synthetic method of 5- methoxyl group -7- azaindole |
| WO2021092262A1 (en) * | 2019-11-05 | 2021-05-14 | Dermira, Inc. | Mrgprx2 antagonists and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008063625A3 (en) | 2008-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008063625A2 (en) | Pyridine compounds and methods of their use | |
| US7297796B2 (en) | Sulfamoyl benzamide derivatives and methods of their use | |
| US7544676B2 (en) | Sulfamoyl benzamides and methods of their use | |
| KR102531491B1 (en) | Modulators of cystic fibrosis transmembrane conduction modulators, pharmaceutical compositions, methods of treatment, and methods of making such modulators | |
| EP2920183B1 (en) | Thieno[3,2-c]pyridin-4(5h)-ones as bet inhibitors | |
| EP2611777B1 (en) | N-(4-{[pyridin-3-yl-methyl)carbamoyl]amino}benzene-sulfone derivatives as nampt inhibitors for therapy of diseases such as cancer | |
| CN109715613A (en) | Heterocyclic compound | |
| CA3133753A1 (en) | Novel small molecule inhibitors of tead transcription factors | |
| UA122391C2 (en) | Isoindoline-1-one derivatives as cholinergic muscarinic m1 receptor positive alloesteric modulator activity for the treatment of alzheimers disease | |
| JPWO2006057270A1 (en) | Nitrogen-containing tricyclic compounds | |
| CA2597410A1 (en) | Substituted morphinans and methods of their use | |
| KR20090110950A (en) | Pyrazine derivatives quinoxaline compounds and use thereof | |
| JP2008138007A (en) | 5-(4-substituted-piperidinyl-1)-3-aryl-pentanoic acid derivative as tachykinin receptor antagonist | |
| TW200808707A (en) | Sulfamide and sulfamate derivatives as histone deacetylase inhibitors | |
| KR20080067656A (en) | Benzamide compounds useful as histone deacetylase inhibitors | |
| JP2008515897A (en) | Phenyl derivatives and methods of using the same | |
| CA3142002A1 (en) | Targeted protein degradation of parp14 for use in therapy | |
| KR20210025631A (en) | Inhibition of CREB binding protein (CBP) | |
| EP2668157A1 (en) | Protease activated receptor 2 (par2) antagonists | |
| CA3139783A1 (en) | Ubiquitin-specific protease inhibitor and preparation method therefor and use thereof | |
| CA2892927A1 (en) | Use of maleimide derivatives for preventing and treating leukemia | |
| KR20240033704A (en) | Derivative based on common anemarrhenae rhizome sarsasapogenin structure, and pharmaceutical composition and use thereof | |
| KR20210141461A (en) | Imidazoquinoline amine derivatives, pharmaceutical compositions, uses thereof | |
| CA2784119A1 (en) | Compounds for the treatment of neurologic disorders | |
| CA2478276A1 (en) | Azepane derivatives and their use as atk1 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07862140 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07862140 Country of ref document: EP Kind code of ref document: A2 |