WO2008027442A2 - Abuse deterrent oral pharmaceutical formulations of opioid agonists and method of use - Google Patents
Abuse deterrent oral pharmaceutical formulations of opioid agonists and method of use Download PDFInfo
- Publication number
- WO2008027442A2 WO2008027442A2 PCT/US2007/019015 US2007019015W WO2008027442A2 WO 2008027442 A2 WO2008027442 A2 WO 2008027442A2 US 2007019015 W US2007019015 W US 2007019015W WO 2008027442 A2 WO2008027442 A2 WO 2008027442A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dosage form
- hours
- oral dosage
- opioid
- opioid agonist
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
Definitions
- the present invention is in the field of abuse deterrent oral pharmaceutical compositions of opioid agonists and the use thereof.
- Nonlimiting examples of agents used include nonsteroidal antiinflammatory agents (NSAIDs), e.g., aspirin, ibuprofen, ketoprofen, diclofenac; opioids, e.g., morphine, hydromorphone, hydrocodone, levorphanol, oxycodone, tramadol, and codeine; cyclooxygenase-2 (COX-2) selective NSAIDs, e.g., celecoxib, valdecoxib, etoricoxib, lumiracoxib, and rofecoxib; acetaminophen; tricyclic antidepressants, e.g., amitriptyline, despiramine, nortriptyline; non-tricyclic antidepressants, e.g., doxepin, duloxetine, paroxetine, venlafaxine
- NSAIDs nonsteroidal antiinflammatory agents
- opioids e.g., morphine, hydromorphone
- opioid agonists such as morphine, hydromorphone, hydrocodone and oxycodone.
- Opioid agonist are powerful analgesics that provided significant relief from virtually all apjnful conditions know to human.
- opioid analgesics are psychoactive substances know to produce iatrogenic addiction in a small nuber of patients with pain. They are also widely sough after by recreational drug users and drug addicts for nonmedical purposes to provide mood altering effects which are perceived to be desireable amongst such recreational drug users and drug addicts.
- a wide variety of pharmaceutical products can be subjected to abuse including opioid agonists.
- NIDA National Institutes of Drag Abuse
- Opioid agonists are currently in widespread medical use and have been associated with a dramatic increase in use by drag addicts and recreational drag users. Both immediate release and extended release opioids can be abused.
- An important goal of analgesic therapy is to achieve continuous relief of pain. Regular administration of an analgesic is generally required to ensure that the next dose is given before the effects of the previous dose have worn off. Continuous suppression of pain through the use of around the clock opioid analgesics is now recommended in treatment guidelines (Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain, Fifth Ed., American, Pain Society (2003); Evidence Based Report of the U.S. Agency for Healthcare Research and Quality (AHRQ) on the Management of Cancer Pain, Report No. 35, AHRQ Publication No. 02-E002, October 2001; Carr et al.
- such formulations can provide more constant plasma concentrations and clinical effects, less frequent peak to trough fluctuations and fewer side effects, compared with short acting opioids (Sloan and Babul, Expert Opinion on Drug Delivery 2006; Babul et al. Journal of Pain and Symptom Management 2004;28:59-71; Matsumoto et al., Pain Medicine 2005;6:357-66; Dhaliwal et al., Journal of Pain Symptom Management 1995;10:612-23; Hays et al., Cancer 1994;74: 1808-16; Arkinstall et al., Painl995;64: 169-78; Hagen et al., Journal of Clinical Pharmacology 1995;35:38-45; Peloso et al., Journal of Rheumatology 2000;27:764-71).
- opioids An important drawback with the use of opioids is the risk of drug addiction, drug diversion and drug abuse. Although the use of opioids for non-medical purposes has existed throughout recorded human history, their abuse has increased significantly in the past two decades (Drug Abuse Warning Network, http://dawninfo.samhsa.gov/; DEA, http://www.deadiversion.usdoj.gov/; National Survey on Drug Use & Health, http://www.oas.samhsa.gov/nhsda.htm; American Association of Poison Control Centers Toxic Exposure Surveillance System, http://www.aapcc.org/annual.htm).
- opioids have been used for non-medical purposes in a variety of settings: i) by patients with pain who have developed an addiction disorder following initiation of opioid therapy; ii) by patients with pain who had a pre-existing addiction disorder; iii) by patients with an addiction disorder seeking opioids for their mood altreing properties.
- Non-medical users of opioid analgesics are generally either recreational drug users who may use such agents episodically, or individuals with a addiction disorder who may require frequent maintenance doses.
- Opioid analgesics may be ingested whole, crushed and ingested, crushed or vaporized and snorted or injected intravenously after attempted extraction of the active pharmaceutical ingredient.
- the manipulation of pharmaceutical dosage forms of opioids has been documented for many decades. For instance, pentazocine (TalwinTM), a synthetic opioid was crushed, extracted and injected intravenously by drug addicts.
- opioid agonists in sustained release form may be crushed or otherwise tampered to liberate make the contents immediate release and then ingested.
- ipioid agonists may be tampered and then administered by the non-oral route (e.g., parenteral, intranasal and inhalations use).
- tampered opioids may be administered with or in the presence of other mood altering licit and illicit mood altering substances and pharmaceutical agents.
- MS ContinTM extended release morphine
- Morphine-3-- glucuronide accumulation has been implicated in hyperalgesia, respiratory stimulation, and behavioral excitatory properties through nonopioid receptor mechanisms. Morphine-6-glucuronide accumulation has been implicated in increasing levels of nausea and sedation in patients with renal impairment (Babul and Darke, Clin Pharm Ther, 1993;54:286-92).
- One mode of abuse involves the extraction of the opioid component from the dosage form by first mixing the table or capsule with a suitable solvent (e.g., water or alcohol), and then filtering and/or extracting the opioid component from the mixture fqr intravenous injection.
- a suitable solvent e.g., water or alcohol
- Another mode of abuse of extended release opioids involves dissolving the drug in water, alcohol or another "recreational solvent" to hasten its release and to ingest the contents orally, in order to provide high peak concentrations and maximum euphoriant effects.
- Winthrop contain a combination of pentazocine and naloxone.
- Pentazocine is a partial agonist at the ⁇ opioid receptors and also has affinity at K opioid receptors, whereas, naloxone is an antagonist of ⁇ receptors.
- Talwin Nx contains pentazocine hydrochloride equivalent to 50 mg base and naloxone hydrochloride equivalent to 0.5 mg base. Talwin Nx is indicated for the relief of moderate to severe pain.
- the amount of naloxone present in this combination has no action when taken orally, and will not interfere with the pharmacologic action of pentazocine. However, this amount of naloxone given by injection has profound antagonistic action to opioid analgesics.
- naloxone is intended to curb a form of misuse of oral pentazocine, which occurs when the dosage form is solubilized and injected. Therefore, this dosage has lower potential for parenteral misuse than previous oral pentazocine formulations.
- ValoronTMN (Goedecke), that comprises tilidine (50 mg) and naloxone (4 mg), has been available in Germany for the management of severe pain.
- U.S. Patent No. 4,457,933 to Gordon et al. teaches the reduction in the oral abuse potential of the analgesics oxycodone, propoxyphene and pentazocine by combining the analgesic with naloxone in a specific range. Naloxone is combined with the selected analgesic a ratio of 2.5-5: 1 part.
- U.S. Patent No. 6,228,863 to Palermo et al. teaches the reduction of the abuse potential of oral dosage forms of opioid analgesics by selecting the particular opioid agonist and antagonist pair, and the concentrations of the same such that the antagonist cannot be easily extracted from the agonist (at least a two-step extraction process being needed to separate the drugs-see also, WO 99/32120).
- the antagonist is in such a concentration that the combination will cause an aversive effect in a physically dependent human subject but not in a naive individual (See also, WO 99/32119).
- U.S. Patent No. 3,773,955 to Pachter et al. describes orally effective analgesic compositions which contain from about 0.1 mg to about 10 mg naloxone with the opioid analgesic. Upon extraction of the composition, parenteral administration is dissuaded, as the dose of naloxone is high enough to prevent the production of analgesia, euphoria or physical dependence from the opioid analgesic.
- WO 01/58447 describes a controlled-release composition which contains an opioid agonist and opioid antagonist that provides an analgesic amount of the opioid agonist over 8 hours along with an amount of opioid antagonist to attenuate a side effect of the opioid agonist.
- WO 01/58451 discloses an oral dosage form comprising an opioid agonist in releasable form and a sequestered opioid antagonist which is substantially not released when the dosage form is administered intact but is released upon tampering.
- WO 99/32120 further describes selecting the opioid agonist and antagonist with respect to physical properties so as to require at least a two-step extraction process to separate the opioid agonist from the antagonist, the amount of opioid antagonist being otherwise sufficient to counteract opioid agonist effect if administered parenterally.
- U.S. Patent No. 4,582,835 to Lewis describes a method of treating pain by administering a sublingually effective dose of buprenorphine with naloxone.
- Lewis describes dosage ratios of naloxone to buprenorphine from 1 :3 to 1:1 for parenteral administration, and from 1:2 to 4: 1 for sublingual administration.
- U.S. Patent No. 6,559,159 to Carroll et al. describes the use of kappa receptors antagonist for the treatment of opioid related addictions.
- One such compound is naltrexone, which is commercially available in the tablet form ReviaTM for the treatment of alcohol dependence and for the blockade of exogenously administered opioids.
- U.S. Patent Nos. 6,277,384, 6,375,957 and 6,475,494 describe oral dosage forms including a combination of an orally active opioid agonist and an orally active opioid antagonist in a ratio that, when delivered orally, is analgesically effective but that is aversive in a physically dependent subject.
- U.S. Patent Nos. 3,980,766, 4,070,494 and 6,309,668 describe formulations designed to prevent the injection of compositions meant for oral administration.
- U.S. Patent No. 3,980,766 describes the incorporation of an ingestible solid which causes a rapid increase in viscosity upon concentration of an aqueous solution thereof.
- U.S. Patent No. 4,070,494 describes the incorporation of a non-toxic, water gelable material in an amount sufficient to render the drug resistant to aqueous extraction.
- U.S. Patent No. 6,309,668 describes a tablet for oral administration containing two or more layers comprising one or more drugs and one or more gelling agents within separate layers of the tablet.
- the resulting tablet forms a gel when combined with the volume of water necessary to dissolve the drug; this formulation thus reduces the extractability of the drug from the tablet.
- these compositions preclude abuse by injection, this approach fails to prevent abuse by crushing and swallowing or snorting the formulation, which are commonly reported methods of abuse associated with OxyContinTM.
- U.S. Patent Nos. 3,773,955 and 3,966,940 describe formulations containing a combination of opioid agonists and antagonists, in which the antagonist does not block the therapeutic effect when the admixture is administered orally, but which does not produce analgesia, euphoria or physical dependence when administered parenterally by an abuser.
- U.S. Patent No. 4,457,933 describes a method for decreasing both the oral and parenteral abuse potential of strong analgesic agents by combining an analgesic dose of the analgesic agent with an antagonist in specific, relatively narrow ratios.
- opioid antagonists themselves have side effects that may be disadvantageous.
- nalorphine causes unpleasant reactions that range from anxiety, to "crazy feelings,” to hallucinations, respiratory depression and miosis.
- Seizures have been reported with naloxone, albeit infrequently, and in postoperative patients, pulmonary edema and ventricular fibrillation have been seen with high dosages.
- Naltrexone has been reported to have the capacity to cause hepatocellular injury when given in doses as low as fivefold or less of therapeutic doses.
- Nalmefene although usually well tolerated, has been reported to cause nausea, vomiting and tachycardia in some individuals.
- opioid antagonists can also precipitate an abstinence syndrome in opioid tolerant patients, resulting in drug withdrawal.
- Symptoms of opioid withdrawal include body aches, diarrhea, gooseflesh, loss of appetite, nervousness or restlessness, runny nose, sneezing, tremors or shivering, stomach cramps, nausea, trouble with sleeping, increased sweating, increased yawning, weakness, increased heart rate or fever. These symptoms can be severe, requiring hospitalization and reinstitution of the opioid agonist.
- Purdue Pharma (Euro-Celtique SA) have reported that one opioid tolerant volunteer among a 24-subject group receiving their extended release opioid agonist with a sequestered opioid antagonist developed severe opioid withdrawal, requiring hospitalization (Sloan and Babul, Expert Opinion on Drug Delivery 2006).
- One novel approach embodied by the present invention involves exploiting the interaction beteeen the cananbinoid and opioidergic systems in initiating and maintaining absue to mood altering systems.
- Another novel approach embodied by the present invention involves modulating the abuse of opioid agonists by exploiting the direct and indirect role of cannabinoid antagonists in suppressing or nullyfing the mood altering effects of the opioid agonist.
- Another novel approach embodied by the present invention involves deterring the abuse of the oral dosage form of the invention by targeting other abusable drugs which are not part of the invention, but which are frequently used, abused or co-abused with opioid agonists (e.g., cannabinoid agonist or alcohol).
- opioid agonists e.g., cannabinoid agonist or alcohol.
- the present invention involves oral dosage forms comprising an opioid agonist and an aversive agent chosen from among cannabinoid antagonists and drugs that produce aversion to alcohol, said aversive agents substantially non- releasable when the dosage form is taken intact and said dosage releasable upon tampering, said dosage form upon tampering effective in reducing or preventing drug abuse, drug diversion, mood alterations desired by drug abusers, said dosage form upon tampering also capable of producing adverse physiological and psychic conseqences.
- cannabinoid antagonist which is sequestered, e.g., is substantially not bioavailable when the dose is administered intact but is bioavailable when the dosage form is tampered with (e.g., in an attempt to misuse the dose of opioid agonist).
- It is a further object of the invention is directed to provide oral pharmaceutical compositions of an opioid agonist which decrease the potential for abuse and co-abuse of cannabinoid agonists that are not part of the dosage form.
- an oral dosage form comprising an opioid agonist and an aversive agent which is present in a substantially non-releasable form (i.e., "sequestered"), said sequestered aversive agent comprising one or more cannabinoid antagonists, one or more alcohol deterrents or a combination thereof.
- the opioid agonists of the present invention can be formulated with a substantially non-releasable aversive agent to deter abuse and/or minimize opioid agonist toxicity on tampering.
- the dosage form contains an orally therapeutically effective amount of the opioid agonist, the dosage form providing a desired analgesic effect. Because the cannabinoid antagonist is present in a substantially non-releasable form, it does not substantially block the therapeutic effects of the opioid agonist when the dosage form is orally administered intact, and does not pose a risk of precipitation of withdrawal in opioid tolerant or dependent patients.
- One novel aspect of the invention concerns deterring or minimizing opioid agonist misuse, abuse and tampering by targeting other co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
- the invention is directed in part to an oral dosage form comprising: (a) an opioid agonist; (b) an aversive agent which is sequestered in the intact dosage form but being releasable upon tampering of said dosage form, such that the intact dosage form releases 49% or less, preferably 42% or less, and more preferably 36% or less, 24.6% or less, 10% or less, or 6.2% or less of the aversive agent after 36 hours based on the in-vitro dissolution of.
- the dosage form in 900 ml of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm and 37 0 C with a switch to Simulated Intestinal Fluid at 1 hour, the aversive agent when released upon tampering of said dosage form at least partially blocking the effect of the opioid agonist and/or at least partially blocking the effect of another abusable drug not included in the dosage form.
- the intact dosage form releases 42% or less, and more preferably 36% or less, 24.6% or less, 10% or less, or 6.2% or less of the aversive agent after 36 hours based on the in-vitro dissolution of the dosage form in 900 ml of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm and 37°C with a switch to Simulated Intestinal Fluid at 1 hour.
- the invention is directed in part to an oral dosage form comprising (a) an opioid agonist in releasable form; (b) an aversive agent in substantially non-releasable form from the intact dosage form but being releasable upon tampering of said dosage form, such that the ratio of the mean Cmax of the aversive agent after single dose oral administration of the dosage form after tampering to the mean Cmax of aversive agent after single dose oral administration of an intact dosage form is at least 1.5: 1.
- said ratio is at least 3: 1 ; or at least 6: 1; or at least 10: 1; or at least 20: 1; or at least 30: 1; or at least 40: 1; or at least 50: 1; or at least 70: 1; or at least 100:1; or at least 500:1.
- the oral dosage form of the present invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4:1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the aversive agent is in the form of multiparticulates individually coated with a sequestering material which substantially prevents release of the aversive agent.
- the oral dosage form of the present invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in- vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the agonist and aversive agent are isolated from each other in two or more distinct layers.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4.1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the aversive agent is dispersed in a matrix comprising a sequestering material which substantially prevents the release of the aversive agent.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 1 hour is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 2 hour is about 4: 1 or greater, based on the in-vitro dissolution at 2 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 3 hour is about 4:1 or greater, based on the in-vitro dissolution at 3 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 4 hour is about 4: 1 or greater, based on the in-vitro dissolution at 4 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 6 hour is about 4:1 or greater, based on the in-vitro dissolution at 6 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of antagonist contained in the intact dosage form to the amount of the antagonist released from the intact dosage form after 8 hour is about 4: 1 or greater, based on the in-vitro dissolution at 8 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of antagonist contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 12 hour is about 4:1 or greater, based on the in-vitro dissolution at 12 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 24 hour is about 4: 1 or greater, based on the in-vitro dissolution at 24 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpra at 37 degrees C.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent, wherein the agonist and aversive agent are interdispersed and visually indistinguishable.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent, wherein the agonist and aversive agent are interdispersed and indistinguishable on the basis of physical characteristics, including bead diameter, density, texture, smell or flotation.
- the invention is directed to an oral dosage form comprising (i) a therapeutic effect of an opioid agonist; and (ii) a sequestered aversive agent, such that at 1 hour after oral administration, the intact dosage form releases not more than about 25% of the aversive agent, the dosage form providing the therapeutic effects of the opioid agonist and the released aversive agent not affecting the therapeutic effects of the agonist, wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers.
- the intact dosage form releases not more than about 12.5% of the aversive agent.
- the invention is directed to an oral dosage form comprising: (i) an intended therapeutic effects of the opioid agonist in a releasable form; and an (ii) aversive agent in substantially non- releasable form wherein the aversive agent is in the form of multiparticulates individually coated with a material which substantially prevents release of the aversive agent.
- the invention is directed to an oral dosage form comprising: (i) an opioid agonist in a releasable form; and a (ii) aversive agent in substantially non-releasable form wherein the aversive agent is dispersed in a matrix comprising a material which substantially prevents the release of the antagonist.
- the invention is directed to an oral dosage form comprising an opioid agonist; and an aversive agent, in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
- the invention is directed to an oral dosage form comprising an opioid agonist; and an orally-bioavailable aversive agent in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
- the invention is directed to an oral dosage form comprising an opioid agonist; and an orally non-bioavailable aversive agent in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
- the invention is directed to an oral dosage form comprising an opioid agonist; and aversive agent with low oral bioavailability in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
- the multiparticulates can be in the form of inert beads coated with the aversive agent and overcoated with a sequestering material, or alternatively in the form of a granulation comprising the antagonist and the material.
- the multiparticulates can be dispersed in a matrix comprising the opioid agonist or contained in a capsule with the opioid agonist.
- the matrix can be in the form of pellets.
- the pellets can be dispersed in another matrix comprising the opioid agonist or contained in a capsule with the opioid agonist.
- the pellets can be visually distinct or visually indistinguishable.
- a portion of the aversive agent is in a matrix and/or part of the aversive agent is in a coated bead.
- the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- the aversive agent is in the form of multiparticulates individually coated with a sequestering material which substantially prevents release of the aversive agent and is subsequently overcoated with the opioid agonist such that each multiparticulate contains an aversive agent and an opioid agonist.
- the aversive agent is in the form of multiparticulates coated with a sequestering material
- the multiparticulates can be in the form of inert beads coated with the aversive agent and overcoated with the sequestering material, and the overcoated still by the opioid agonist, or alternatively in the form of a granulation comprising the antagonist and the sequestering material, and the overcoated still by the opioid agonist.
- the multiparticulates can be dispersed in a matrix and compressed into a tablet or contained in a capsule.
- the intact dosage form releases 20% or less of the aversive agent after 1 hour and the tampered dosage form releases 80% or more aversive agent after 1 hour.
- the intact dosage form releases 10% or less of said aversive agent after 1 hour and the tampered dosage form releases 40% or more aversive agent after 1 hour.
- the intact dosage form releases 5% or less of said aversive agent after 1 hour and the tampered dosage form releases 20% or more aversive agent after 1 hour.
- the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of said aversive agent released from the intact dosage form based on the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. is 10: 1 or greater, 50: 1 or greater or 100: 1 or greater.
- the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of said aversive agent released from the intact dosage form based on the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. is 7.5: 1 or greater, 15: 1 or greater, 30: 1 or greater or 60:1 or greater.
- the aversive agent in a substantially non-releasable form is adapted to release less than 15% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 8% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 3% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non- releasable form is adapted to release less than 1% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 0.5% by weight in vivo after 36 hours.
- the invention is also directed to methods of preventing abuse of an opioid agonist utilizing the dosage forms disclosed herein.
- the invention is also directed to methods of preventing abuse of a cannabinoid agonist utilizing the dosage forms disclosed herein, said agonist not part of the dosage form, said agonist used concurrently or contemporaneously with the dosage form.
- the invention is also directed to methods of preventing abuse of alcohol utilizing the dosage forms disclosed herein, said alcohol not part of the dosage form, said alcohol used concurrently or contemporaneously with the dosage form.
- the invention is also directed to methods of preventing abuse of a opioid agonists, cannabinoid agonists and/or alcohol in the setting of polydrug abuse or poly-substance abuse utilizing the dosage forms disclosed herein.
- the method for the foregoing can comprise providing the opioid agonist in an oral dosage form together with an aversive agent, wherein the aversive agent is present in a form which is substantially non-releasable form oral ingestion when the integrity of the dosage form is maintained at the time of oral ingestion, but which becomes bioavailable or releasable if subjected to tampering (e.g., crushing, grinding, pulverizing, heating, solvent immersion, solvent extraction, followed by oral, parenteral, intranasal or inhalational use or abuse).
- tampering e.g., crushing, grinding, pulverizing, heating, solvent immersion, solvent extraction, followed by oral, parenteral, intranasal or inhalational use or abuse.
- Another embodiment of the invention is directed to a method of decreasing the abuse of an opioid agonist in an oral dosage form, comprising preparing an oral dosage form as disclosed herein.
- the method can comprise preparing a dosage form which comprises (i) an orally therapeutically effective amount of an opioid agonist and (ii) an aversive agent in a substantially non-releasable form such that said dosage form provides a desired therapeutic effect and said antagonist does not substantially block the effect of the aversive agent when said dosage form is administered orally intact.
- the effect of the opioid agonist is at least partially blocked when said dosage form is tampered with(e.g., crushing, grinding, pulverizing, heating, solvent immersion, solvent extraction, followed by oral, parenteral, intranasal or inhalational use or abuse).
- the invention is also directed to a method of treating or preventing diseases and disorders amenable to treatment with opioid agonists with the dosage forms disclosed herein.
- the method can comprise providing an oral dosage form containing an opioid agonist in a releasable form and an aversive agent in substantially non-releasable form; and orally administering the intact oral dosage form.
- Another embodiment of the invention is directed to a method of preventing or treating pain with the disclosed dosage forms.
- the method of treating pain in patients with a dosage form having less abuse potential comprises providing an oral dosage form containing a releasable form of an opioid agonist and a substantially non- releasable form of an aversive agent; and orally administering the oral dosage form to provide a blood plasma level of agonist greater than the minimum analgesic concentration of the opioid agonist.
- Another embodiment of the invention is directed to a method of preventing or treating diseases and disorders amenable to treatment with opioid agonists with the disclosed dosage forms.
- the method of preventing or treating such diseases and disorders in patients with a dosage form having less abuse potential comprises providing an oral dosage form containing a releasable form of an opioid agonist and a substantially non- releasable form of an aversive agent; and orally administering the oral dosage form to provide a blood plasma level of agonist greater than the minimum analgesic concentration of the opioid agonist.
- the invention is also directed to methods of preparing the dosage forms disclosed herein.
- the invention comprises a method of preparing an oral dosage form comprising pretreating an aversive agent to render it substantially non-releasable; and combining the pretreated aversive agent with a releasable form of opioid agonist in a manner that maintains the integrity of the non-releasable form of the aversive agent.
- the one or more aversive agents in sequestered (i.e., non-releasable or substantially releasable) form are chosen from the group comprising cannabinoid antagonists and alcohol deterrents.
- Certain embodiments of the invention are directed to formulations wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers. However in certain embodiments, the agonist and aversive agent are partially interdispersed.
- the dosage form comprises one or more opioid agonists in releasable or substantially releasable form; and one or more aversive agents in a non-releasable or substantially releasable form when said dosage form is used as intended.
- the dosage form comprises one or more opioid agonists in releasable or substantially releasable form, and one or more cannabinoid antagonists and one or more opioid antagonists, each in a non-releasable or substantially releasable form, when said dosage form is used as intended.
- the dosage form comprises one or more opioid agonists in releasable or substantially releasable form, and one or more cannabinoid antagonists plus one or more alcohol deterrents, each in a non-releasable or substantially releasable form, when said dosage form is used as intended.
- the dosage form comprises one or more opioid agonists in releasable or substantially releasable form, and one or more opioid antagonist plus one or more alcohol deterrents, each in a non-releasable or substantially releasable form, when said dosage form is used as intended.
- Mammalian tissues express at least two cannabinoid receptors, both of which are G-protein coupled. These are CBj receptors and CB 2 receptors. CBj receptors are expressed are primarily expressed in peripheral and central nerve terminals where they mediate inhibition of neurotransmitter release. In the CNS, especially high levels of CBi receptors are found in the cerebellum, hippocampus and basal ganglia. CB 2 receptors are found primarily on immune and hematopoietic cells outside (and also within) the central nervous system, where they appear to modulate cytokine release and immune cell migration.
- CBi and CB 2 receptor knockout mice indicate that some of the effects of endocannabinoids are not mediated by either CB i or CB 2 receptors, suggesting the existence of additional yet to be identified sites of action. Some cannabinoid effects resist classification as either CB) and CB 2 -mediated. There is growing evidence suggesting the involvement of additional receptors, which include TRPVi receptors and at least 2 G protein-coupled receptors (GPCRs) of unclear molecular identity that have only been defined pharmacologically.
- GPCRs G protein-coupled receptors
- the human cannabinoid system is involved in a number of pathological states, including Alzheimer's disease, schizophrenia, depression, alcoholism, Parkinson's disease, stroke, premature labor, endotoxic shock, hepatic cirrhosis, atherosclerosis, cancer, bone implantation, glaucoma, emesis and pain.
- upregulation or downregulation of the endocannabinoid system is seen in a variety of animal in vivo models, including multiple sclerosis, amyotrophic lateral sclerosis, encephalitis, Alzheimer's disease, Parkinson's disease, Huntington's disease, pain, obesity, feeding, fasting, stress, memory, aging, hypertension, cirrhosis, septic shock, cardiogenic shock, cerebral ischemia, myocardial infarction, neurotoxicity, febrile seizures and various intestinal disorders.
- cannabinoid receptors present a large number of potential targets for pharmacologic intervention and efforts are underway to develop and test a variety of cannabinoid agonists and antagonists to prevent and treat various maladies.
- three non-specific cannabinoid receptor agonists are commercially available.
- Nabilone (CesametTM) and dronabinol (MarinolTM) are oral synthetic THC analogs which have been shown effective for the treatment of nausea and vomiting associated with cancer chemotherapy and AIDS-related cachexia.
- a buccal spray containing THC and cannabidiol (SativexTM) was approved in Canada for the symptomatic relief of neuropathic pain in multiple sclerosis.
- Cannabinoid agonists can produce a variety of adverse effects including ⁇ a number of psychotomimetic effects such as dizziness, drowsiness, euphoria, ataxia, anxiety, disorientation, depression, hallucinations, vertigo, and psychosis. While these psychic effects are undesirable for patients, they are often sought after by recreational drug users and individuals with an addiction disorder. Cannabinoids play a modulatory role in drug seeking. They can reinstate cocaine seeking behavior after several weeks of extinction of intravenous cocaine self-administration. Similar effects have been shown in animals with a history of heroin, methamphetamine, alcohol and nicotine self-administration where cannabinoid receptor agonists have reinstated previously abolished drug seeking.
- Addiction to drugs is characterized by long-lasting motivational disturbances including compulsive drug seeking, intense drug craving, use despite harm, the non-medical use and diversion of psychoactive substances, manipulation of the medical system and escalating drug use and risk taking behaviors.
- the neurobiological mechanisms underlying such behaviors are poorly understood.
- Cannabinoids play a modulatory role in drug seeking.
- cannabinoid agonists and opioid agonists activate mu, delta and kappa opioid, and CB], CB 2 and non-CB]/CB 2 cannabinoid receptors, respectively, which are coupled to Gi/Go GTP-binding proteins that inhibit adenylyl cyclase, inhibit voltage-dependent calcium channels, stimulate potassium channels and activate the MAP kinase cascade (for review see Childers, 1991; Childers et al., 1992; Howlett, 1995).
- cannabinoid agonists and opioid agonists results in pharmacologic tolerance, physical dependence and addiction.
- Chronic cannabinoid agonist administration induces tolerance to the antinociceptive effect of opioids (Smith et al., 1994; Welch, 1997), while chronic exposure to opioid agonists results in tolerance to the antinociceptive effect of cannabinoid agonists (Bloom and Dewey, 1978; Hine, 1985; Smith et al., 1994; Thorat and Bhargava, 1994).
- Cannabinoid agonists and opioid agonists seem to interact in their antinociceptive effects as illustrated by the ability of their respective antagonists to reverse cannabinoid/opioid-induced analgesia (Welch, 1993; Reche et al., 1996a,b; Cichewicz et al., 1999).
- the concurrent administration of opioid agonsits and cannabinoid agonists results in an enhanced antinociceptive effect, compared with either solo administartion (Cichewicz et al., 1999; Smith et al., 1998; Welch and Eads, 1999; Cichewicz and McCarthy, 2003).
- the reward process is central to the development of addiction to psychoactive drugs.
- a commonly used experimental method of evaluating the reinforcing properties of drugs is the self-administration test. Available data suggest that there is an an interaction between opioids and cannabinoids with respect to reward processes.
- the cannabinoid antagonist SR141716A reduces self-administration of heroin (Chaperon et al., 1998; Braida et al., 2001; Mas- Nieto et al., 2001; Navarro et al., 2001; De Vries et al., 2003).
- the opioid antagonists naltrexone and naloxone reduce self-administration of THC (Tanda et al., 2000; Justinova et al., 2003, 2004) and the CBl agonist CP- 55,940 (Braida et al., 2001).
- Cannabinoid antagonists can also suppress "heroin-seeking" behavior after weeks of prior extinction (Fattore et al., 2003; Caille and Parsons, 2003; Solinas et al., 2003).
- a majority of opioid-dependent individuals seeking treatment are polydrug abusers.
- the secondary licit or illicit abusable or mood altering drug used most frequently in this population is marijuana and alcohol.
- Prevalence estimates of marijuana use have ranged from 25% to 80% among cocaine and opioid agonist abusers (Ball et al., 1988; Budney et al., 1996;Miller et al., 1990; Nirenberg et al., 1996; Saxon et al.,1993).
- Budney et al (Addiction, 1998) evaluated marijuana use among opioid abusers in patients enrolled in treatment for opioid dependence. Sixty-six per cent of participants were current marijuana users and almost all (94%) continued to use during treatment.
- opioid agonist users are polydrug abusers.
- a frequently co-abused substance in this population is alcohol.
- One novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target the role of cannabinoid antagonist in nullifying the mood altering effects of opioid agonists.
- Another novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target the role of cannabinoid antagonist in producing an aversive effects and in precipitating ari abstinence syndrome (withdrawal syndrome) in subjects tolerant to opioid agonists.
- Yet another novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target other co-consumed or co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
- opioid agonists which deter abuse by: (i) providing an aversive effect towards co-consumed or co-abused mood altering substances such as alcohol and cannabinoid agonists which are not part of the dosage form; (ii) nullifying the mood altering effects of the abused opioid agonist; and/or (iii) providing an aversive or adverse effect directed at the opioid agonist and any co-consumed opioid agonists or alcohol.
- the present invention relates to oral opioid agonist in releasable form and aversive agents in substantially non-releasable form (i.e., sequestered), said dosage forms having reduced potential for abuse in opioid agonist abusers and in polydrug abusers.
- the invention Under conditions of abuse or tampering (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered aversive agent (e.g., alcohol deterrent or cannabinoid antagonist) on: (i) co-used or co-abused alcohol or cannabinoid agonist, particularly in the setting of polydrug abuse and (ii) nullification of the mood altering effects of the opioid agonist.
- the sequestered aversive agent e.g., alcohol deterrent or cannabinoid antagonist
- the present invention is directed to immediate and controlled release oral dosage form of opioid agonists which, are formulated in order to reduce and minimize misuse, abuse and diversion.
- opioid agonists which, are formulated in order to reduce and minimize misuse, abuse and diversion.
- these characteristics are conferred by the inclusion of a cannabinoid antagonist, which is itself formulated in a unique controlled release matrix.
- the properties of this formulation are developed to liberate the cannabinoid antagonist in conditions of misuse or tampering yet a negligible amount of cannabinoid antagonist would be released (an amount which does not affect therapeutic effect of the opioid agonist experienced by the patient) under the prescribed conditions of use.
- the present invention is based in part on several observations: (i) opioid agonists (or opioid analgesics) are frequently abused in the setting of polydrug abuse (e.g., co-abuse with marijuana or alcohol) and (ii) the endocannabinoid system interacts with the opioidergic system. These observations can be exploited to deter opioid agonist abuse by including in the opioid agonist dosage form a substantially non-releasable cannabinoid antagonist in quantities sufficient to nullify the effects of the abused opioid agonist and/or the co-used or co-abused cannabinoid agonist, said cannabinoid agonist not part of the dosage form.
- the invention provides a method for preventing abuse of a oral opioid agonist dosage form where the dosage form also includes a dose of cannabinoid antagonist or alcohol deterrent which is sequestered, e.g., is substantially not bioavailable when the dose is administered intact but is bioavailable when the dosage form is tampered with (e.g., in an attempt to misuse the dose of opioid agonist).
- the invention to provide an oral dosage form of an opioid agonist and a substantially non-releasable cannabinoid antagonist that is useful for decreasing the potential abuse of the opioid agonist when administered intact, without affecting the therapeutic effects of the opioid agonist or incurring the risk of precipitating signs and symptoms of opioid agonist withdrawal.
- the sequestered cannabinoid agonist upon tampering becomes substantially releasable and produces its abuse and misuse deterrence by nullifying some or all of the mood altering effects of the cannabinoid agonist co-abused by the subject or by precipitating an abstinence syndrome (i.e., signs and symptoms of cannabinoid agonist withdrawal) in subjects with cannabinoid agonist tolerance and physical dependence.
- an abstinence syndrome i.e., signs and symptoms of cannabinoid agonist withdrawal
- the sequestered cannabinoid antagonists upon tampering releases some or a substantial amount of its cannabinoid antagonist content from the dosage form of the invention, thereby nullifying some or a substantial amount of the mood altering effect of the opioid agonist, as well as any co-abused cannabinoid agonist that subject has taken in the past, is taking concurrently or is expected to take in the near future (e.g., cannabinoid agonist use within minutes, hours or days of use of the tampered dosage form of the invention).
- the sequestered cannabinoid antagonists upon tampering releases some or a substantial amount of its cannabinoid antagonist content from the dosage form of the invention, thereby precipitating an opioid agonist abstinence syndrome (i.e., signs and symptoms of opioid agonist withdrawal), as well as a cannabinoid agonist abstinence syndrome (i.e., signs and symptoms of cannabinoid agonist withdrawal), in subjects with opioid agonist and cannabinoid agonist tolerance and physical dependence, respectively.
- opioid agonist abstinence syndrome i.e., signs and symptoms of opioid agonist withdrawal
- a cannabinoid agonist abstinence syndrome i.e., signs and symptoms of cannabinoid agonist withdrawal
- the ratio of the opioid agonist to the substantially non-releasable form of the aversive agent in the oral dosage form is such that the effect of the opioid agonist is at least partially blocked when the dosage form is chewed, crushed or dissolved in a solvent and heated, and administered orally, intranasally, inhalationally, parenterally or sublingually.
- the oral dosage form of the present invention when administered properly as intended, would not substantially release the cannabinoid antagonist, the amount of such antagonist may be varied more widely than if the cannabinoid antagonist is available to be released into the gastrointestinal system upon oral administration. For safety reasons, the amount of the antagonist present in a substantially non-releasable form should not be permanently harmful to humans even if fully released.
- the ratio of particular cannabinoid agonist to antagonist can be determined by one skilled in the art.
- the ratio of the opioid agonist to the aversive agent, present in a substantially non-releasable form is about 1:10,000 to about 10000:1 by weight, or about 1 :1000 to about 1000: 1 by weight or preferably about 1 : 10 to about 10: 1 by weight, and more preferably about 5: 1 to 1:5 by weight.
- the weight ratio of the opioid agonist to aversive agent, as used in this application, refers to the weight of the active ingredients.
- analgesic effectiveness is defined for purposes of the present invention as a satisfactory prevention, reduction in or elimination of pain, along with a tolerable level of side effects, as determined by the human patient.
- therapeutic effectiveness is defined for purposes of the present invention as a satisfactory prevention or treatment of diseases and disorders amenable to treatment with an opioid agonist, including their signs and symptoms, along with a tolerable level of side effects, as determined by the human patient.
- An "agonist” is a ligand that binds to a rebeptor and alters the receptor state resulting in a biological response. Conventional agonists increase receptor activity, whereas inverse agonists reduce it (See Neubig et al, IUPHAR Committee on Receptor Nomenclature and Classification, Pharmacol Rev, 2003; Howlett et al., MoI Pharmacol, 1988).
- An "antagonist” is a drug of ligand that reduces the action of another drug or ligand, generally an agonist. Many antagonists act at the same receptor macromolecule as the agonist. (See Neubig et al, IUPHAR Committee on Receptor Nomenclature and Classification, Pharmacol Rev, 2003; Howlett et al., MoI Pharmacol, 1988).
- receptor means a molecule within a cell, on a cell surface, on a membrane, in tissue, in fluid or otherwise found in humans that serves as a recognition or binding site to cause specific physiologic, pathophysiologic or pharmacologic effects.
- the term “receptor” also means a cellular macromolecule, or an assembly of macromolecules, that is concerned directly and specifically in chemical signaling between and within cells. Combination of a hormone, neurotransmitter, drug, ligand, or intracellular messenger with its receptors) initiates a change in cell function (Neubig et al, IUPHAR Committee on Receptor Nomenclature and Classification, Pharmacol Rev, 2003).
- a cannabinoid antagonist in a substantially non-releasable form refers to said agent that is not released or substantially not released at one hour after the intact dosage form containing both opioid agonist and the said agent is orally administered (i.e., without having been tampered with).
- the amount released after oral administration of the intact dosage form may be measured in-vitro via the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
- a dosage form is also referred to as comprising a "sequestered aversive agent", or a “sequestered agent”, or as applicable, a “sequestered cannabinoid antagonist", or as applicable, a “sequestered alcohol deterrent”.
- tampering means any manipulation including by mechanical, thermal and/or chemical means which changes the physical properties of the dosage form, e.g., to liberate the opioid for immediate release if it is in sustained release form, or to make the opioid agonist available for inappropriate use such as administration by an alternate route, e.g., parenterally inhalationally, intranasally.
- the tampering can be, e.g., by means of crushing, shearing, grinding, chewing, dissolution in a solvent, heating, mechanical extraction, solvent extraction, solvent immersion, combustion, or any combination thereof.
- the term "at least partially blocking the [e.g., cannabinoid agonist or opioid agonist or alcohol] effect” is defined for purposes of the present invention to mean that the cannabinoid antagonist at least partially or at least significantly blocks the mood altering effects of the opioid agonist, or the cannabinoid agonist or alcohol taken separately by the human subject or patient, thereby reducing the potential for abuse of the opioid agonist in the dosage form.
- microood altering is defined for purposes of the present invention to mean that the "high”, “liking”, pleasurable, euphoric, calming, anxiolytic, auditory and visual perceptual alterations, relaxing, psychotomimetic, mood altering, rewarding, reinforcing alterations in perception, cognition and mental focus; sexual gratification; sexual arousal; sexual desire and sexual anticipation; increased socialization effects of the abusable drug.
- abuse means single use, intermittent use, repeated use, recreational use and chronic use of the specified abusable drug or class of abusable drugs: (i) in quantities or by methods and routes of administration that do not conform to standard medical practice; (ii) outside the scope of specific instructions for use provided by a qualified medical professional; (iii) outside the supervision of a qualified medical professional; (iv) outside the approved instructions on proper use provided by the drug's legal manufacturer; (v) which is not in specifically approved dosage forms for medical use as pharmaceutical agents; (vi) where there is an intense desire for and efforts to procure same; (vii) compulsive use; (viii) through acquisition by manipulation of the medical system, including falsification of medical history, symptom intensity,
- “deter abuse” “, “deter misuse”, resist abuse” and “resist misuse” and “deter abuse” are used interchangeably in the context of the present invention and include pharmaceutical compositions and methods that resist, deter, discourage, diminish, delay and/or frustrate: (i) the intentional, unintentional or accidental physical or chemical manipulation or tampering of the dosage form (e.g., crushing, shearing, grinding, chewing, dissolving, melting, needle aspiration, inhalation, insufflation, extraction by mechanical, thermal and chemical means, and/or filtration); (ii) the intentional, unintentional or accidental use or misuse of the dosage form outside the scope of specific instructions for use provided by a qualified medical professional, outside the supervision of a qualified medical professional and outside the approved instructions on proper use provided by the drug's legal manufacturer (e.g., intravenous use, intranasal use, inhalational use and
- aversive agents means to compounds contained within the dosage form that produce an aversive, undesirable, repugnant, distasteful, unpleasant, unacceptable physiologic or unacceptable psychic effects, or alcohol deterrence, or that pharmacologically block or reduce the mood altering effects of the dosage form or of concurrently or contemporaneously used abusable drugs, preferably selected from the group comprising cannabinoid agonists, alcohol of combinations thereof.
- sustained release is defined for purposes of the present invention as the release of the cannabinoid agonist from the oral dosage form at such a rate that blood (e.g., plasma) concentrations (levels) are maintained within the therapeutic range (above the minimum effective concentration) but below toxic levels over a period of 4 to 24 hours, preferably over a period of time indicative of a rwice-a-day or a once-a-day formulation.
- blood e.g., plasma
- concentrations levels
- levels levels
- subject for purposes of treatment is used interchangeably with “patient”, “male”, “female”, and includes any human who has a medical condition amenable to prevention or treatment with an opioid agonist.
- pathological states are used interchangeably and are intended to have their broadest interpretation to refer to any physiologic, pathologic or pathophysiologic state in a human, including the signs and symptoms thereof that can be prevented, treated, managed or altered to produce a desired, usually beneficial effect.
- pharmaceutical agent pharmaceutical agent
- pharmaceutical agent pharmaceutical agent
- active agent active agent
- agent agent
- agent agent
- “Pharmaceutically or therapeutically acceptable excipient or carrier” refers to a solid or liquid filler, diluent or encapsulating substance which does not interfere with the effectiveness or the biological activity of the cannabinoid agonist and which is not toxic to the hosts, which may be either humans or animals, to which it is administered.
- salts refers to a salt which is toxicologically safe for human and animal administration.
- Nonlimiting examples of salts include hydrochlorides, hydrobromides, hydroiodides, sulfates, bisulfates, nitrates, citrates, tartrates, bitartrates, phosphates, malates, maleates, napsylates, fumarates, succinates, acetates, terephlhalates, pamoates and pectinates.
- the xenobiotic pharmaceutical composition is a salt or complex of inorganic cation salts, organic salts such- primary, secondary, tertiary and quaternary amines include substituted amines
- suitable pharmaceutically acceptable salts of xenobiotic include any of the inorganic cation salts such as sodium, potassium, lithium, magnesium, calcium, cesium, ammonia, ferrous, zinc, manganous, aluminum, ferric, and manganic; organic salts with primary, secondary, tertiary and quaternary amines, or mixtures thereof.
- Examples of such primary, secondary, tertiary and quaternary amines include substituted amines including but not limited to naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and mixtures thereof. More specifically, suitable amines include but are not limited to tromethamine, triethylamine, tripropylamine, dropopizine, 2-dimethylaminoethanol, 2- diethylaminoethanol, lysine, arginine, ornithine, histidine, caffeine, procaine, N-ethylpiperidine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, fr ⁇ -(hydroxymethyl)aminomethane, ⁇ -methylglucamine, methylglycamine, theobromine, piperazine, piperidine, polyamine resins and the like, and mixtures thereof.
- suitable amines include but are not limited to tromethamine, triethylamine,
- examples of suitable pharmaceutically acceptable salts include aminoalcohols chosen from the group consisting of ethanolamine, 3-amino-l-propanol, (7?)-l-amino-2-propanol, (5)-l-amino- 2-propanol, 2 -amino- 1 ,3-propandiol, N-(2-hydroxyethyl)pyrrolidine, D-glucamine and L-prolinol, D-glucosamine, and N-methylglucosamine.
- aminoalcohols chosen from the group consisting of ethanolamine, 3-amino-l-propanol, (7?)-l-amino-2-propanol, (5)-l-amino- 2-propanol, 2 -amino- 1 ,3-propandiol, N-(2-hydroxyethyl)pyrrolidine, D-glucamine and L-prolinol, D-glucosamine, and N-methylglucosamine.
- examples of suitable pharmaceutically acceptable salts include alkali and alkaline earth metals and salts of an organic nature, such as the salts of basic amino acids.
- Some of the opioid agonists, cannabinoid antagonists and alcohol deterrents disclosed herein may contain one or more asymmetric centers and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms.
- the present invention is also meant to encompass all such possible forms as well as their racemic and resolved forms and mixtures thereof.
- the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended to include both E and Z geometric isomers. All tautomers are intended to be encompassed by the present invention as well.
- stereoisomers is a general term for all isomers of individual molecules that differ only in the orientation of their atoms is space. It includes enantiomers and isomers of compounds with more than one chiral center that are not mirror images of one another (diastereomers).
- chiral center refers to a carbon atom to which four different groups are attached.
- enantiomer or “enantiomeric” refers to a molecule that is nonsuperimposeable on its mirror image and hence optically active wherein the enantiomer rotates the plane of polarized light in one direction and its mirror image rotates the plane of polarized light in the opposite direction.
- racemic refers to a mixture of equal parts of enantiomers and which is optically inactive.
- Aversive agents of the oral dosage form of the invention comprise one or more cannabinoid agonists, one or more alcohol deterrents or mixtures there of, said aversive agent(s) in the dosage form in substantially non-releasable form. When the dosage form is tampered, the aversive agent becomes partially or substantially releasable.
- the aversive agents are in the form of pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- the amount of substantially non-releasable aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists and alcohol deterrents may be about 10 ng to 1500 mg, more preferably, 10 ng to 1000 mg, even more preferably 0.1 mg to 800 mg, and most preferably, 0.1 mg to 500 mg.
- the amount of aversive agent required to produce the desired aversive effect will vary, but can be readily determined based on the physicochemical and pharmaceutical properties and the pharmacology of the drug, including its oral bioavailability, half life, intrinsic clearance, potency, absolute bioavailability, potency, safety and the like, and the magnitude of desired aversive effect.
- cannabinoid agonist means a substance that binds to one or more cannabinoid receptor to exert an agonist or partial agonist effect.
- cannabinoid antagonist means an antagonist substance or an inverse agonist that binds to one or more cannabinoid receptor to exert an antagonist effect.
- cannabinoid receptor means a molecule that causes a specific physiologic, pathophysiologic or pharmacologic effect after binding to CBi, CB 2 , non-CBi/CB 2 cannabinoid sites, TRPVi receptors, as well as other G protein-coupled receptors (GPCRs) that form part of the endocannabinoid system (Wiley and Martin, Chemistry Physics of Lipids, 2002; Begg et al., Pharmacol Ther, 2005; Howlett et al., Neuropharmacol, 2004; Pertwee, AAPS Journal, 2005; International Union of Pharmacology (IUPHAR) Receptor Database; Howlett et al., MoI Pharmacol, 1988).
- GPCRs G protein-coupled receptors
- Cannabinoid agonists are known or readily determined by individuals who practice the art.
- the cannabinoid agonist useful for the present invention may be selected from the group consisting of inhibitors of cannabinoid agonist metabolism (e.g., without limitation, URB602, an inhibitor of monoacylglycerol lipase which catalyzes 2-arachidonoylglycerol hydrolysis) THC, nabilone, dronabinol, cannabidiol, 9-THC propyl analog, cannabidiol, cannabidiol propyl analog, cannabinol, cannabichromene, cannabichromene propyl analog, cannabigerol, cannabinoid terpenoids, cannabinoid flavonoids, endocannabinoids, anandamide, (R)- methanandamide,and 2-arachidonoylglycerol, THC-like ABC tricyclic cannabinoid an
- the cannabinoid agonist useful for the present invention may be selected from the group consisting of dexanabinol (HU211), BAY 38-7271 , Naphthalen- 1 -yl-(4-pentyloxynaphthalen- 1 -yl)methanone, THC (delta-9-tetrahydrocannabinol), nabilone, dronabinol, cannabidiol, cannabinol, cannabichromene, cannabigerol, cannabigerol, anandamide, (R)- methanandamide, 2-arachidonoylglycerol, HU210, desacetyllevonantradol, CP55940, CP55244, URB602, or WIN55212-2 and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates
- the cannabinoid agonist useful for the present invention may be selected from the group consisting of 9-THC propyl analog, endocannabinoids, cannabinoid terpenoids, cannabinoid flavonoids, inhibitors of cannabinoid agonist metabolism, inhibitors of monoacylglycerol lipase, cannabidiol propyl analogues, cannabichromene propyl analogues, THC-like ABC tricyclic cannabinoid analogues, synthetic AC bicyclic cannabinoid analogues, synthetic ACD tricyclic cannabinoid analogues, aminoalkylindole compounds or analogs of 2-Arylimino-5,6-dihydro-4H-l,3-thiazines and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual
- the amount of the cannabinoid agonist used by the subject may be from about 10 ng to about 2000 mg, even up to about 2000 mg. More preferably, the amount of the cannabinoid agonist is from about 10 ng to about 1500 mg, even more preferably from about 0.1 mg to about 1000 mg, and most preferably, from about 0.1 mg to about 700 mg.
- the cannabinoid agonist may be selected from compounds disclosed in U.S. Patent No. 7,217,732, 7,214,716, 7,169,942, 7,109,216, 7,091,216, 7,057,051 , 6,995, 184, 6,972,295, 6,943,266, 6,903, 137, 6,864,291, 6,864,285, 6,525,087, 6,524,805, 6.509.367. 6,284,788, 5,948,777, 5,939,429, and 5,605,906, and in U.S. Patent Application No.
- cannabinoid agonist shall include combinations of more than one cannabinoid agonist, and also include the unsalif ⁇ ed agonist, mixed agonist-antagonists, partial agonists, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
- cannabinoid agonist for the purposes of the present invention, 1) drugs that enhance the effect of cannabinoid agonists by inhibiting their metabolism or reuptake (for example, anandamide amidase inhibitors) are considered to be cannabinoid agonists; 2) drugs that induce anandamide amidase inhibitor metabolism or induce CBj, CB 2 and non-CB
- the cannabinoid antagonist useful for the present invention may be selected from the group consisting of SR 141716A [Rimonabant or N-piperidino-5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-4-methyl-3-pyrazole-carboxamide]), AM251, AM 281 ([N- mo ⁇ holin-4-yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3- carboxamide), AM630, SR 144528 ([N-[(lS)-endo- 1,3,3- trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3-methyl-phenyl)- 1 -(4- methylbenzyl)pyrazole-3 -carboxamide]), 5-(4-chlorophenyl)- 1 -(2,4- dichlorophenyl)-3 -
- cannabinoid antagonist shall include combinations of more than one cannabinoid agonist, and also include the unsalified agonist, mixed agonist-antagonists, partial agonists, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
- the amount of the cannabinoid antagonist in the composition may be from about 10 ng to about 1000 mg, even up to about 2000 mg. More preferably, the amount of the cannabinoid antagonist is from about 10 ng to about 1200 mg, even more preferably from about 0.1 mg to about 1000 mg, and most preferably, from about 0.1 mg to about 700 mg.
- the cannabinoid antagonist may be selected from compounds disclosed in U.S. Patent No. 7,247,628, 7,176,210, 7,153,997, 7, 151,097, 7,132,414, 7,1 19, 108,6,930, 122, 6,642,258, 7,094,794, 6,916,838, 6,894,050, 6,875,782, 6,825, 198, 6,734,176, 6,673,802, 6,630,507, 6,555,578, 6,509,367, 6,344,481, 6,344,474, 6,194,454, 6,100,259, 5,989,583, 5,939,429, 5,747,524, 5,596,106, and 4,205,952, and in U.S.
- cannabinoid antagonist shall include combinations of more than one cannabinoid antagonist, and also include the unsalified antagonist, mixed agonist- antagonists, partial agonists, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
- cannabinoid antagonists block the euphoric, pleasurable, reinforcing or toxic effects of the cannabinoid agonist. Furthermore, it is known that cannabinoid antagonist administration in the setting of dependence or pharmacologic tolerance to cannabinoid agonists results in aversive effects, which may include signs and symptoms of cannabinoid agonist withdrawal. [00195] In certain embodiments, the effect of the cannabinoid agonist is at least partially blocked by the cannabinoid antagonist. In certain other embodiments, the effect of the cannabinoid agonist is substantially blocked by the cannabinoid antagonist.
- the cannabinoid antagonists precipitates signs or symptoms cannabinoid agonist withdrawal or abstinence in individuals who have developed tolerance to the cannabinoid agonist. In certain embodiments, the cannabinoid antagonist precipitates aversive effects of cannabinoid agonist withdrawal which discourage future abuse of the dosage form.
- a frequently co-abused substance in recreational drug users and drug addicts, including heroin users is alcohol.
- One novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target other co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
- alcohol deterrent or “alcohol deterrence” means a molecule that causes a specific adverse physiologic, pathophysiologic or pharmacologic adverse effect to a human in the presence of alcohol or reduces the desire for alcohol.
- All kinds of aversive effects of the alcohol deterrent in the presence of alcohol are anticipated, including a reduced desire for alcohol, nullification of mood altering and pleasurable effects, cutaneous flushing, vasodilation, throbbing, headache, respiratory distress, nausea, vomiting, retching, sweating, thirst, chest pain, hypotension, orthostatic hypotension, dizziness, syncope, anxiety, uneasiness, weakness, vertigo, blurred vision, confusion, facial flush, pallor and/or shock.
- the alcohol deterrent when the dosage form is tampered, the alcohol deterrent precipitates aversive effects of alcohol which discourages future abuse of the dosage form containing the cannabinoid agonist.
- the alcohol deterrent may also block the mood altering, euphoric, pleasurable, reinforcing, rewarding or toxic effects of the cannabinoid agonist upon tampering of the dosage form.
- Alcohol deterrents are known or readily determined by individuals who practice the art. Alcohol deterrents include disulfiram, calcium carbimide, acmaprosate, diethylthiomethylcarbamate, inhibitors of aldehyde dehydrogenase, metabolites of disulfiram, metronidazole, chlorpropamide, topiramate, opioid receptor antagonists including naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan nalorphine, nalmefene, nadide, levallorphan, N-methylnaltrexone, N- allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N- allyllevallorphan.
- the oral opioid agonist is in releasable form and the formulation further includes as an aversive agent an alcohol deterrent(s), said dosage forms having reduced potential for abuse in polydrug abusers.
- the invention does not provide abuse deterrence from the sequestered alcohol deterrent of the dosage form on the releasable opioid agonist of the dosage form under conditions of normal use (when administered whole or intact).
- the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered alcohol deterrent on: (i) co-used or co-abused alcohol, particularly in the setting of polydrug abuse; (ii) nullification of the mood altering effects of the opioid agonist in the presence of alcohol.
- the amount of the alcohol deterrent in the claimed composition may be from about 10 ng to about 1000 mg, even up to about 2000 mg. More preferably, the amount of the alcohol deterrent is from about 10 ng to about 1200 mg, even more preferably from about 0.1 mg to about 1000 mg, and most preferably, from about 0.1 mg to about 700 mg.
- a frequently co-abused substance in recreational drug users and drug addicts, including heroin users is cannabis.
- One novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target other co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
- Cannabinoid agonist abuse in the setting of polydrug abuse from the tampering of immediate release and particularly extended or sustained release formulations can be minimized by combining the releasable opioid agonist with a non-releasable or substantially non-releasable (i.e., sequestered) cannabinoid antagonist in the same dosage form, such that upon tampering, the cannabinoid antagonist becomes releasable, thereby: (i) reducing or eliminating the psychic effects of any co-abused cannabinoid agonist desired drug addicts and recreational drug users; and (ii) reducing or eliminating the toxic effects of the combination of the opioid agonist and the co-abused cannabinoid agonist in drug addicts and recreational drug users.
- a non-releasable or substantially non-releasable i.e., sequestered
- opioid receptor includes mu ( ⁇ ), delta ( ⁇ ), kappa (K) and/or nociceptin/orphanin FQ (N/OFQ) peptide (NOP) receptors, their subtypes and splice variants such as ⁇ i, ⁇ 2 , ⁇ i, ⁇ 2 , Ki, K 2 and K 3 , etc, regardless of whether they also bind to or influence other receptor systems (e.g., norepinephrine reuptake inhibition, serotonin reuptake inhibition, NMDA receptor antagonism).
- NOP nociceptin/orphanin FQ peptide
- opioid is interchangeable with the term “opioid agonist”, except when there is a specific reference to an opioid antagonist.
- Opioid agonists include alfentanil, allylprodine, alphaprodine, anileridine, apomorphine, apocodeine, benzylmorphine, bezitramide, brifentanil, buprenorphine, butorphanol, carfentanil, clonitazene, codeine, cyclo ⁇ hen, cyprenorphine, desomo ⁇ hine, dextromoramide, dezocine, diampromide, dihydrocodeine, dihydromo ⁇ hine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxyaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmo ⁇ hine, etonitazene, fentanyl, heroin, hydrocodone, hydroxymethylm
- Opioid agonists also include drugs that bind to opioid receptors to exert agonist activity and are listed in the United States Controlled Substances Act of 1970, as amended, and regulations thereof, and drugs listed in the United States Psychotropic Substances Act of 1978, as amended, and regulations thereof.
- opioid antagonist or "opioid receptor antagonist” means an antagonist substance that binds to one or more opioid receptor to exert an antagonist effect.
- Opioid antagonists are known or readily determined by individuals who practice the art.
- the opioid antagonists useful for the present invention may be selected from the group consisting of naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan nalorphine, nalmefene, nadide, levallorphan, N-methylnaltrexone, N- allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N- allyllevallorphan.
- the ratio of the opioid agonist and the cannabinoid antagonist, present in a substantially non- releasable form is about 1: 1000 to about 1000: 1 by weight or about 1: 1000 to about 1000: 1, or about 1: 100 or about 100: 1 by weight, preferably about 1 : 10 to about 10:1 by weight, and more preferably about 5: 1 to 1:5 by weight.
- the present invention provides an oral dosage form of opioid agonist useful for decreasing the potential for abuse of the opioid agonist contained therein.
- the present invention includes an oral dosage form comprising an orally therapeutically effective amount of an opioid agonist in combination with a cannabinoid antagonist.
- the cannabinoid antagonist is present in a substantially non-releasable form'.
- the present invention relates to oral opioid agonist in releasable form and cannabinoid antagonist in substantially non- releasable form (i.e., sequestered) when administered intact, said dosage forms having reduced potential for abuse in opioid abusers and polydrug abusers.
- the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered cannabinoid antagonist on co-abused cannabinoid agonists in the setting of polydrug abuse under conditions where the dosage form of the invention is abuse or tampered (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), said cannabinoid agonist not part of the dosage form and said dosage form having no significant direct effect on the opioid agonist of the dosage form when the dosage form is used as directed or, in some embodiments, even when subject to abuse, and on any co-used cannbinoid agonist when the dosage form of the invention is not abused or tampered.
- a novel method namely by the effects of the sequestered cannabinoid antagonist on co-abused cannabinoid agonists in the setting of polydrug abuse under conditions where the dosage form of the invention is abuse or tampered (e.g., crushing or solvent extraction, followed by in
- the present invention relates to oral opioid agonist in releasable form and an alcohol deterrent in substantially non- releasable form (i.e., sequestered) when administered intact, said dosage forms • having reduced potential for abuse in alcohol abusers and polydrug abusers.
- the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered alcohol deterrent on used or co-abused alcohol in the setting of polydrug abuse under conditions where the dosage form of the invention is abused or tampered (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), said alcohol not part of the dosage form and said dosage form having no significant direct effect on the opioid agonist of the dosage form when the dosage form is used as directed or, in some embodiments, even when subject to abuse in the absence of alcohol, and on any co-used alcohol when the dosage form of the invention is not abused or tampered.
- a novel method namely by the effects of the sequestered alcohol deterrent on used or co-abused alcohol in the setting of polydrug abuse under conditions where the dosage form of the invention is abused or tampered (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), said
- the cannabinoid antagonist present in a substantially non-releasable form does not substantially block the therapeutic effects of the opioid agonist when the dosage form is orally administered intact (and does not pose a risk of precipitation of withdrawal in cannabinoid tolerant or dependent patients), but wherein the effect of the opioid agonist is at least partially blocked by the cannbinoid antagonist when said dosage form is tampered with, e.g., chewed, crushed or dissolved in a solvent, and administered orally, intranasally, by inhalation, parenterally or sublingually.
- the substantially non-releasable form of the aversive agent comprises aversive agent particles in a coating that substantially prevents the release of the aversive agent.
- the coating comprising one or more of pharmaceutically acceptable hydrophobic material.
- the coating is preferably impermeable to the cannabinoid antagonist contained therein and is insoluble in the gastrointestinal system, thus substantially preventing the release of the cannabinoid antagonist when the dosage form is administered orally as intended.
- the preferred embodiments of the invention comprise a cannabinoid antagonist or alcohol deterrent in a form that completely prevents the release of the aversive agent
- the invention also includes an aversive agent in a substantially non-releasable form.
- the term "substantially not released” N refers to the aversive agent that might be released in a small amount, as long as the amount released does not affect or does not significantly affect therapeutic efficacy of the agonist when the dosage form is orally administered to humans as intended.
- the substantially non-releasable form of the aversive agent is resistant to laxatives (e.g., mineral oil) used to manage constipation.
- laxatives e.g., mineral oil
- the substantially non-releasable form of the aversive agent is formulated with one or more of pharmaceutically acceptable hydrophobic material, such that the antagonist is not released or substantially not released during its transit through the gastrointestinal tract when administered orally as intended, without having been tampered with.
- the substantially non- releasable form of the aversive agent is vulnerable to mechanical, thermal and/or chemical tampering, e.g., tampering by means of crushing, shearing, grinding, chewing, and/or dissolution in a solvent in combination with heating of the oral dosage form.
- mechanical, thermal and/or chemical tampering e.g., tampering by means of crushing, shearing, grinding, chewing, and/or dissolution in a solvent in combination with heating of the oral dosage form.
- the dosage form when the dosage form is chewed, crushed or dissolved and heated in a solvent, and administered orally, intranasally, inhalationally, parenterally or sublingually, the analgesic, euphoric, pleasurable, reinforcing or toxic effects of the cannabinoid is reduced or eliminated.
- the hydrophobic material comprises a cellulose polymer or an acrylic polymer that is insoluble in the gastrointestinal fluids and impermeable to the aversive agent.
- aversive agent particles refers to granules, spheroids, beads or pellets comprising the aversive agent.
- the aversive agent particles are about 0.2 to about 2 mm in diameter, more preferably about 0.5 to about 2 mm in diameter.
- the oral dosage form further comprises an aversive agent (e.g., cannabinoid antagonist or alcohol deterrent) in a releasable form and is thus capable of being released from the oral dosage form when orally administered, the ratio of the opioid agonist to the releasable form of the aversive agent (e.g., cannabinoid antagonist) being such that the dosage form, when administered orally, is therapeutically effective as an opioid agonist.
- an aversive agent e.g., cannabinoid antagonist or alcohol deterrent
- aversive agent e.g., cannabinoid antagonist
- a coating that substantially prevents its release and is then mixed with an opioid agonist and compressed into tablets
- certain amounts of the coating might be cracked, thus exposing the aversive agent (e.g., cannabinoid antagonist) to be released upon oral administration.
- the ratio of the opioid agonist to the aversive agent e.g., a cannabinoid antagonist, or an alcohol deterrent
- the aversive agent e.g., a cannabinoid antagonist, or an alcohol deterrent
- the ratio of the opioid agonist to the aversive agent is about 10000: 1 to about 1: 10000, or about 1000:1 to about 1: 1000 by weight, or preferably about 1 : 100 to 100: 1 by weight or about 1 : 10 to about 10: 1 by weight, and more preferably about 5: 1 to 1:5 by weight.
- the weight ratio of the opioid agonist to the aversive agent refers to the weight of the active ingredients.
- the weight of the aversive agent excludes the weight of the coating or matrix that renders the aversive agent substantially non-releasable, or other possible excipients associated with the aversive agent particles. Since the aversive agent is in a substantially non- releasable from, the amount of such aversive agent within the dosage form may be varied more widely, as the formulation does not depend on differential biotransformation or pharmacodynamics for proper functioning. For safety reasons, the amount of the aversive agent present in a substantially non- releasable form is selected as not to be permanently harmful to humans even if fully released by tampering with the dosage form.
- the oral dosage form of the present invention is directed to an oral dosage form comprising (i) a opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the mean C max of the aversive agent after single dose oral administration of the dosage form after tampering to the mean C max of aversive agent after single dose oral administration of an intact dosage form is at least 1.5: 1.
- the mean C max ratio using the aforementioned test method is at least 3: 1, 6: 1, 10: 1, 20: 1, 30: 1, 40:1, 50: 1, 70: 1, 100: 1 or 500: 1.
- the ratio of the mean AUCo -t or AUCo-oo of the aversive agent after single dose oral administration of an immediate release reference product containing an equivalent amount of aversive agent to the mean AUCo -1 or AUCo- ⁇ of aversive agent after single dose oral administration of an intact dosage form is at least 1.5:1.
- the mean AUCo-t or AUCo- ⁇ ratio using the aforementioned test method is at least 3: 1, 6: 1, 10: 1, 20:1, 30: 1, 40: 1, 50: 1, 70: 1, 100: 1 or 500: 1.
- the ratio of the mean Tmax of the aversive agent after single dose oral administration of the intact dosage form to the mean Tmax of aversive agent after single dose oral administration of a dosage form after tampering is at least 1.5: 1.
- the mean Tmax ratio using the aforementioned test method is at least 3: 1, 6: 1, 10: 1 or 20: 1.
- the oral dosage form containing a opioid agonist in combination with a substantially non-releasable form of an aversive agent includes, but are not limited to tablets or capsules.
- the dosage forms of the present invention may include any desired pharmaceutical excipients known to those skilled in the art.
- Specific examples of pharmaceutically acceptable carriers and excipients that may be used to formulate oral dosage forms of the present invention are described in the Handbook of Pharmaceutical Excipients, APhA Publications; 5 edition (January 5, 2006), compounds found on the FDA EAFUS database (http://vm.cfsan.fda.gov/ ⁇ dms/eafus.html); FDA Food Additives Status List (http://www.cfsan.fda.gov/ ⁇ dms/opa-appa.html); FDAGRAS list and database; FDA Color Additive Status List
- the oral dosage forms may further provide an immediate release of the opioid agonist.
- the oral dosage forms of the present invention provide a sustained release of the opioid agonist contained therein.
- Oral dosage forms providing sustained release of the opioid agonist may be prepared in accordance with formulations/methods of manufacture known to those skilled in the art of pharmaceutical formulation, e.g., via the incorporation of a sustained release carrier into a matrix containing the substantially non-releasable form of an aversive agent; or via a sustained release coating of a matrix containing the opioid agonist and the substantially non-releasable form of the aversive agent.
- the benefits of the abuse-resistant dosage form are especially great in connection with oral dosage forms of potent opioid agonists, which can provide valuable therapeutic benefits but are prone to being abused. This is particularly true for sustained release opioid agonist products which have a large dose of a desirable opioid agonist intended to be released over a period of time in each dosage unit. Drug abusers take such sustained-release product and crush, grind, extract or otherwise damage the product so that the full contents of the dosage form become available for immediate absorption. Since such tampering of the dosage form of the invention results in the cannabinoid antagonist also becoming available for absorption, the present invention provides a-means for deterring such abuse. In addition, the present invention addresses the risk of overdose to ordinary patients from "dumping" effect of the full dose of the opioid agonist if the product is accidentally chewed or crushed.
- the invention may provide for a safer product (e.g., lower risk of opioid agonist toxicity or polydrug toxicity), if the product is misused, as well as one with less risk of abuse.
- a safer product e.g., lower risk of opioid agonist toxicity or polydrug toxicity
- a combination of two opioid agonists is included in the formulation with the aversive agent.
- one or more opioid agonist and an aversive agent is included and a further non-opioid agonist drug is also included for the treatment of the same medical condition as the opioid agonist or for the treatment of a different medical condition.
- a combination of two or more aversive agents for interfering with the same or a different type of abuse e.g., two cannabinoid antagonists; a cannabinoid antagonist and an alcohol deterrent are included in the formulation with the opioid agonist(s).
- the dosage form is co-administered with a non-opioid agonist drug for the treatment of the same medical condition as the opioid agonist or for the treatment of a different medical condition.
- All modes of co-administration are contemplated, including via oral, subcutaneous, direct intravenous, slow intravenous infusion, continuous intravenous infusion, intravenous or epidural patient controlled analgesia (PCA and PCEA), intramuscular, intrathecal, epidural, intracisternal, intramuscular, intraperitoneal, transdermal, topical, transmucosal, buccal, sublingual, transmucosal, inhalation, intranasal, epidural, intra-atricular, intranasal, rectal or ocular routes.
- PCA and PCEA patient controlled analgesia
- oral pharmaceutical dosage forms of the invention are contemplated, including oral suspensions, tablets, capsules, lozenges, effervescent tablets, transmucosal films, buccal products, oral mucoretentive products and the like, administered as immediate release, sustained release, delayed release, modified release, controlled release, extended release and the like.
- the dosage form may include, in addition to the releasable opioid agonist and the substantially non-releasable cannabinoid antagonist, other abuse deterrent substances in releasable or substantially non- releasable form, including various aversive agents know to practitioners of the art.
- a variety of pharmaceutically-acceptable carriers may be used.
- Nonlimiting examples are sugars, starches, cellulose and its derivatives, malt, gelatin, talc, calcium sulfate, vegetable oils, synthetic oils, polyols, alginic acid, phosphate buffered solutions, emulsifiers, isotonic saline, and pyrogen-free water.
- Other pharmaceutically active ingredients (drugs) from various therapeutic classes may also be used in combination with the present invention, e.g., included in the dosage forms of the invention. They include, but are not limited to decongestants, analgesics, analgesic adjuvants, antidepressants, antipsychotics, anxiolytics, hypnotics, sedatives, anti-ADHD drugs, psychostimulants, drugs to treat urinary incontinence, antihistamines, expectorants, antitussives, diuretics, anti-inflammatory agents, antipyretics, antirheumatics, antioxidants, laxatives, local anesthetics, proton pump inhibitors, motility modifying agents, vasodilators, inotropes, beta blockers, beta adrenergic agonists, drugs to treat asthma and COPD, antiinfectives, antimigraine agents, antihypertensives, antianginal agents, gastric acid reducing agents, anti-ulcer
- the drug being used in combination therapy with the present invention can be administered by any route, including parenterally, orally, topically, transdermally, sublingually, and the like.
- the invention allows for the use of lower doses of the opioid agonist by virtue of the inclusion of an additional non-opioid agonist drug for the prevention or treatment of the same medical condition. By using lower amounts of either or both drugs, the side effects associated with treatment in humans are reduced.
- the present invention is further directed to a method of decreasing the potential for abuse of an opioid agonist in an oral dosage form. The method comprises providing the opioid agonist in an oral dosage form as described herein.
- the present invention is directed to an immediate and controlled release opioid agonists which are formulated in order to reduce and minimize misuse, abuse and diversion.
- these characteristics are conferred by the inclusion of a cannabinoid antagonist or alcohol deterrent, which is itself formulated in a unique controlled release matrix.
- the properties of this formulation are developed to liberate the aversive agent in conditions of misuse or tampering yet a negligible amount of aversive agent would be released (an amount which does not affect therapeutic effect of the opioid agonist experienced by the patient) under the prescribed conditions of use.
- the release for the aversive agent (e.g., antagonist component) of the formulation is expressed in terms of a ratio of the release achieved after tampering, e.g., by crushing or chewing, relative to the amount released from the intact formulation.
- the ratio is therefore expressed as [Crushed]/[Whole], and it is desired that this ratio have a numerical range of at least 4: 1 or greater (crushed release in 1 hour/intact release in 1 hour).
- the present invention provides an oral dosage form of opioid agonist useful for decreasing the potential for abuse of the opioid agonist contained therein.
- the present invention includes an oral dosage form comprising an orally therapeutically effective amount of an opioid agonist in combination with an aversive agent (e.g., cannabinoid antagonist).
- an aversive agent e.g., cannabinoid antagonist.
- the cannabinoid antagonist is present in a substantially non-releasable form.
- the aversive agent in a substantially non-releasable form comprises aversive agen particles coated with a coating that substantially prevents its release.
- a coating surrounds the aversive agent particles and is impermeable to the drug and is insoluble in the gastrointestinal system.
- the aversive agent e.g., cannabinoid antagonist
- the dosage form of the present invention is orally administered to humans, the aversive agent (e.g., cannabinoid antagonist) is not substantially released from the coating and is, therefore, not available for absorption into the body.
- the aversive agent e.g., cannabinoid antagonist
- the aversive agent e.g., cannabinoid antagonist
- the oral dosage form of the present invention is tampered with as to compromise the integrity of the coating, the cannabinoid antagonist contained therein would be made available to at least partially block the effect of the opioid agonist.
- This characteristic decreases the potential for abuse or diversion of the opioid agonist in the oral dosage form.
- a solvent with heat e.g., greater than about 45. degree. C. to about 50. degree.
- the coating will be damaged and will no longer prevent the cannabinoid antagonist from being released.
- the aversive agent e.g., cannabinoid antagonist
- the aversive agent will be released and significantly block the euphoric, pleasurable, reinforcing or toxic effects of the opioid agonist and of any co-used or co-abused cannabinoid agonist.
- the ratio of the opioid agonist to the coated aversive agent is such that when the oral dosage form is tampered with as to compromise the integrity of the coating that renders the aversive agent (e.g., cannabinoid antagonist) substantially non-releasable, the euphoric, pleasurable, reinforcing or toxic effects of the agonist would be negated by the cannabinoid antagonist when misused by a human subject orally, parenterally, intranasally, inhalationally or sublingually.
- the aversive agent e.g., cannabinoid antagonist
- the euphoric, pleasurable, reinforcing or toxic effects of the opioid agonist would be negated by the aversive agent (e.g., cannabinoid antagonist) when misused parenterally or sublingually.
- the aversive agent e.g., cannabinoid antagonist
- the present invention also includes an oral dosage form which comprises a releasable form of an averdive agent (e.g., cannabinoid antagonist), along with a opioid agonist and coated aversive agent (e.g., cannabinoid antagonist) particles, the ratio of the agonist to the non-coated aversive agent (e.g., cannabinoid antagonist) being such, when administered orally as intended, the oral dosage form is therapeutically or analgesically effective.
- an averdive agent e.g., cannabinoid antagonist
- a opioid agonist and coated aversive agent e.g., cannabinoid antagonist
- the aversive agent in a substantially non-releasable form comprises an aversive agent (e.g., cannabinoid antagonist) dispersed in a matrix that renders the antagonist substantially non-releasable, wherein the matrix comprises one or more of a pharmaceutically acceptable hydrophobic material.
- the aversive agent e.g., antagonist
- the aversive agent is substantially not released from the matrix, thus is not made available to be absorbed during its transit through the gastrointestinal system.
- the aversive agent e.g., cannabinoid antagonist
- aversive agent e.g., cannabinoid antagonist
- the matrix comprises one or more of a pharmaceutically acceptable hydrophobic material.
- the oral dosage form of the present invention may further include, in addition to a opioid agonist and aversive agent (e.g., antagonist), one or more drugs that may or may not act synergistically therewith.
- a combination of two opioid agonists may be included in the dosage form, in addition to the aversive agent (e.g., cannabinoid antagonist).
- the dosage form may include two opioid agonists having different properties, such as half-life, solubility, potency, and a combination of any of the foregoing.
- one or more opioid agonist is included and a further non- opioid agonist drug is also included, in addition to the aversive agent (e.g., cannabinoid antagonist).
- a non-opioid agonist drug is also included for the treatment of the same medical condition as the opioid agonist or for a different medical condition.
- the opioid agonist is intended to prevent or treat acute or chronic pain.
- An included non- opioid agonist drug in such a dosage form may be used to provide additive, complementary, or synergistic therapeutic effects, including NSAIDs, NO-NSAIDs, COX-2 selective inhibitors, acetaminophen, nitroparacetamol, nitric oxide donors, tramadol, beta adrenergic agonists, alpha-2 agonists, selective prostanoid receptor antagonists, cannabinoid agonists, opioid receptor agonists, NO-opioid receptor agonists, local anesthetics, purinergic P2 receptor antagonists, NMDA receptor antagonists, gabapentin, pregabalin, gabapentinoids, ligands of alpha(2)delta subunits of voltage-gated calcium channels, neuronal nicotinic receptor agonists, calcium channel antagonists, sodium channel blockers, superoxide dismutase mimetics, p38 MAP kinase inhibitors, TRPVl agonists, dextrome
- the dosage form optionally comprises, in addition to the foregoing opioid agonist and the aversive agent(s) (selected from cannabinoid antagonists and alcohol deterrents, and combinations of the same), one of more additional agents that are referred to herein as an abuse intervention agent(s), in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form.
- aversive agent(s) selected from cannabinoid antagonists and alcohol deterrents, and combinations of the same
- an abuse intervention agent(s) selected from cannabinoid antagonists and alcohol deterrents, and combinations of the same
- the abuse intervention agent(s) may comprise, for example, laxatives, cutaneous vasodilators, headache producing agents, emetics, emetogenic compound, nausea producing compounds, bittering agents, drugs that cause burning on irritation when in contact with tissue or mucous membranes (e.g., naso-mucosal irritants, oro-mucosal irritants, respiratory irritants), tissue irritants, gastrointestinal irritants, drugs that precipitate withdrawal effects, tissue dyes, lakes and colorants, beverage dyes, lakes and colorants, non-tissue staining beverage dyes, lakes and colorants (i.e, that do not stain or discolor the skin upon ingestion), fecal discolorants, urine discolorants, malodorous agents, opioid antagonists, benzodiazepine antagonists (e.g., flumazenil), and the like.
- laxatives e.g., cutaneous vasodilators, headache producing agents, emetics
- the abuse intervention agents are further selected from the group comprising (i) laxatives; (ii) cutaneous vasodilators; (iii) headache producing agents; (iv) emetics, emetogenic and nausea producing compounds; (iv) bittering agents (v) mucosal, naso-mucosal, oro-mucosal, respiratory, tissue and gastrointestinal irritants; (vi) tissue staining, non-tissue staining and beverage staining dyes, lakes and colorants; (vii) fecal and urine discolorants; (viii) malodorous agents; (ix) opioid antagonists; and (x) benzodiazepine antagonists (e.g., flumazenil), and mixtures thereof.
- laxatives e.g., cutaneous vasodilators; (iii) headache producing agents; (iv) emetics, emetogenic and nausea producing compounds; (iv) bittering agents (v) mucosal
- the abuse intervention agent comprises a non-toxic dye to deter surreptitious attempts at intoxication of another subject (e.g., in an alcoholic or nonalcoholic beverage).
- the dosage form comprises an abuse intervention agent which comprises a nontoxic bittering agent to deter surreptitious attempts at intoxication of another subject (e.g., in an alcoholic or non-alcoholic beverage).
- an abuse intervention agent which comprises a nontoxic bittering agent to deter surreptitious attempts at intoxication of another subject (e.g., in an alcoholic or non-alcoholic beverage).
- the dosage form comprises an abuse intervention agent which comprises a nontoxic bittering agent to deter oral or nasal ingestion of the dosage form.
- the dosage form comprises an abuse intervention agent which comprises a nontoxic nasal irritant to deter oral or nasal ingestion of the dosage form.
- the abuse intervention agent(s) may be in the dosage form in an amount that does not produce an aversive effect or aversion in any, many or substantially all patients when taken in accordance with the prescribing information or the manufacturer's instructions (for example, in small quantities), but which produce an aversive effect when taken in excess (e.g., higher dose or more frequently).
- the abuse intervention agent is one or more bittering agents selected from the group comprising T2R or TAS2R receptor agonists, phenylthiourea (phenylthiocarbamide), natural, artificial and synthetic flavor oils, flavoring aromatics, flavoring oils, oleoresins, spearmint oil, peppermint oil, eucalyptus oil, oil of nutmeg, allspice, mace, oil of bitter almonds, menthol, citrus oils including lemon, orange, lime, grapefruit, and fruit essences, sucrose derivatives, sucrose octaacetate, chlorosucrose derivatives, quinine, denatonium, denatonium saccharide and denatonium benzoate.
- bittering agents selected from the group comprising T2R or TAS2R receptor agonists, phenylthiourea (phenylthiocarbamide), natural, artificial and synthetic flavor oils, flavoring aromatics, flavoring oils, oleoresins, spearmint oil, pepper
- the abuse intervention agent is one or more naso-mucosal, oro-mucosal, respiratory or tissue irritants selected from the group comprising transient receptor potential vanilloid 1 agonists, resiniferanoids, capsaicinoids, phorboid vanilloids, terpenoid 1,4-unsaturated dialdehydes, capsaicin, capsaicin analogs, resiniferatoxin, olvanil, pipeline, zingerone, anandamide, 12- and 15-(S)-hydroperoxy-eicosatetraenoic acids, 5 -•and 15-(S)-hydroxyeicosatetraenoic acids, phorbol 12-phenylacetate 13- acetate 20-homovanillate, 2 phorbol 12,13-didecanoate 20-homovanillate, leukotriene B(4), tinyatoxin, heptanoylisobutylamide, N-
- the abuse intervention agent is one or more emetogenic or nausea producing agents selected from the group comprising zinc and pharmaceutically acceptable salts thereof, dopamine agonists, apomorphine, ipecac, ipecacuanha, emetine, methylcephaeline, cephaeline, psychotrine, O-methylpsychotrine, ammonium chloride, potassium chloride, magnesium sulfate, ferrous gluconate, ferrous sulfate, aloin, algarot or antimonious oxychloride, antimony trichloride, folate, folic acid, niacin and nicotinamide.
- emetogenic or nausea producing agents selected from the group comprising zinc and pharmaceutically acceptable salts thereof, dopamine agonists, apomorphine, ipecac, ipecacuanha, emetine, methylcephaeline, cephaeline, psychotrine, O-methylpsychotrine, ammonium chloride, potassium chloride
- the abuse intervention agent is one or more cutaneous vasodilators selected from the group comprising niacin, nicotinuric acid, beta-hydroxybutyrate and nicotinic receptor agonists, including agonists at nicotinic receptor HM74A and nicotinic receptor GPR 109 A.
- the abuse intervention agent is one or more tissue dyes, lakes or colorants, or beverage dyes, lakes or colorants, or a beverage dye, lake and colorant that does not stain or discolor the skin upon ingestion, or a fecal discolorant or a urine discolorant selected from the group comprising Curcumin, Riboflavin, Tartrazine, Quinoline yellow, Sunset yellow FCF, Carmine, Carmoisine, Amaranth, Ponceau 4R, Erythrosine, Allura red AC, Patent blue V, Indigo carmine, Brilliant blue FCF, Chlorophylls, Copper complexes of chlorophylls and chlorophyllins, Green S, Caramel, Brilliant black BN, Vegetable carbon, Carotenoids, Alpha-, beta-, gamma-carotene, Capsanthin, Capsorubin, Lycopene, Beta-apo-8' carotenal, Ethyl ester of beta-apo-8' caro
- the abuse intervention agent is one or more laxatives selected from the group comprising Bis(p-hydroxyphenyl)pyridyl-2- methane, bisacodyl, bisoxatin, anthraquinone, anthraquinone analogs and derivatives (e.g., buckthorn, casanthranol, cascara, hydroxyanthracene, glucofrangulin ), dantron, danthron, docusate (e.g., docusate sodium, docusate calcium, docusate potassium), gastrointestinal chloride channel activators (e.g., chloride channel subtype 2 activators), lubiprostone, magenesium salts (e.g., magnesium citrate, magnesium hydroxide, magnesium oxide), mannitol, oxyphenisatine, polyethylene glycol, polyethylene oxide) [PEO- 1500], sodium phosphate, phenolphthalein, senna, senna constituents and
- the abuse intervention agent is one or more opioid antagonists selected from the group comprising naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan nalorphine, nalmefene, nadide, levallorphan, N-methylnaltrexone, N- allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N- allyllevallorphan.
- the abuse intervention agent may be added to the formulation in an amount of less than about 80% by weight, preferably less than about 60% by weight, more preferably less than about 40% by weight of the dosage form, even more preferably less than about 20% by weight of the dosage form, and most preferably less than about 10 by weight of the dosage form (e.g., 0.000000000000001% to 1%, or 0.000000001% to 3%, or 0.0001% to 10%, or 0.001% to 5%, or 1% to 10%, or 0.001% to 2%, or 1% or 10%, or 2% to 7%) depending on the particular aversive agent used.
- the abuse intervention agent in the dosage form may be about 0.00000000001 mg to about 2000 mg, or about 0.0000001 mg to about 1500 mg, or about 0.000001 mg to about 1000 mg, or about 0.0001 mg to about 1000 mg, or about 0.001 mg to about 1000 mg, or about 0.01 mg to about 1000 mg, or about 0.1 mg to about 1500 mg, or 1 mg to about 800 mg, or about 1 mg to about 500 mg, or about 1 mg to about 300 mg, or about 1 mg to about 150 mg, or about 5 mg to about 400 mg, or about 5 mg to about 200 mg, or about 0.00000000001 mg to about 200 mg, or about 0.00000000001 mg to about 100 mg, or about 0.00000000001 mg to about 50 mg, or about 0.0000001 mg to about 200 mg, or about 0.0000001 mg to about 100 mg, or about 0.00001 mg to about 400 mg, or about 0.0001 mg to about 300 mg.
- the amount of the abuse intervention agent in the dosage form of the present invention can be a fixed ratio in relation to the amount of opioid agonist in the dosage form.
- aversive effects can be avoided under conditions of proper medical use (e.g., manufacturers prescribing directions).
- the quantity of aversive agent consumed will exceed the "no effect” or "minimum effect” threshold, thereby producing one or more aversive effects, for example, e.g., nausea, emesis, diarrhea, laxation, cutaneous vasodilation, headache, bitter taste, naso-mucosal irritation, oro-mucosal irritation, reduction of the pleasurable, mood altering, rewarding, reinforcing, or other psychic and physiologic effects of the opioid agonist or a co-abused drug.
- aversive effects for example, e.g., nausea, emesis, diarrhea, laxation, cutaneous vasodilation, headache, bitter taste, naso-mucosal irritation, oro-mucosal irritation, reduction of the pleasurable, mood altering, rewarding, reinforcing, or other psychic and physiologic effects of the opioid agonist or a co-abused drug.
- the "no effect" or “minimum effect” threshold amount of the abuse intervention agent can be exceeded when the dosage form of the invention is taken in excess of the manufacturer's recommendation by a factor of about 1.5, or about 2, or about 2.5, or about 3, or about 4, or about 5, or about 6, or about 7, or about 8, or about 10, or more than 10.
- the production of an aversive effect can reduce or stop further abuse of the dosage form, thereby reducing the harm or toxicity of the drug in the subject who is tampering, misusing or abusing the dosage form, e.g., addicts, drug abusers and recreational drug users.
- the opioid agonist of the invention may be used for the prevention or treatment of any diseases and disorders, including without limitation, (i) pain; (ii) infectious, immunologic, cardiovascular, pulmonary, gastrointestinal, hepatic, biliary, nutritional, metabolic, endocrine, hematologic, oncologic, musculoskeletal, rheumatic, neurologic, psychiatric, genitourinary, gynecologic, obstetric, pediatric, otolaryngogologic, ophthalmic, dermatologic, dental, oral, and genetic disorders, diseases and maladies and signs and symptoms thereof; (iii) depression, schizophrenia, influenza, common colds, anxiety, panic attacks, agoraphobia, ADHD, insomnia, sleep disorders, nasal congestion, headaches, migraine, urinary incontinence, constipation, allergies, cough, pneumonia, COPD, asthma, fluid retention, acid reflux, peptic ulcers, hypertension, cardiac arrhythmias, hypercholesterolemia, CHF, fever, diarrhea
- peripheral neuropathic pain e.g., acute and chronic inflammatory demeyelinating polyradiculopathy, alcoholic polyneuropathy, chemotherapy-induced polyneuropathy, complex regional pain syndrome (CRPS) Type I and Type II, entrapment neuropathies (e.g., carpal tunnel syndrome), HIV sensory neuropathy, iatrogenic neuralgias (e.g., postthoracotomy pain, postmastectomy pain), idiopathic sensory neuropathy, painful diabetic neuropathy, phantom limb pain, postherpetic neuralgia, trigeminal neuralgia, radiculopathy (e.g., cervical thoracic, lumbosacral), sciatica, acute herpes zoster pain, temporomandibular joint disorder pain and postradiation plexopathy; and (ii) central neuropathic pain, e.g., peripheral neuropathic pain, e.g., acute and chronic inflammatory demeyelinating polyradiculopathy, alcoholic poly
- Meaningful Pain Relief are assessed and defined as follows: At the time of dosing with the study medication, a trained member of study staff starts two stopwatches for each patient. The patient is instructed to stop the first stopwatch at the time of perceptible pain relief and the second stopwatch at the time when they first experience meaningful pain relief.
- the usual definitions of the perceptible and meaningful pain relief are as follows: Perceptible Pain Relief is when the patient begins to feel any pain relieving effect from the drug. The patient is typically instructed as follows: "I would like you to stop the first stopwatch when you first feel any pain relief whatsoever. This does not mean you feel completely better, although you might, but when you first feel any difference in the pain that you have had”. Meaningful Pain Relief is when the patient feels their pain relief is meaningful to them.
- the patient is typically instructed as follows: "I would like you to stop the second stopwatch when you have meaningful pain relief. That is, when the relief from the pain is meaningful to you". Confirmed Perceptible Pain Relief is Perceptible Pain Relief in those patients who go on to also have Meaningful Pain Relief.
- This questionnaire can be used to examine the overall drug effects of abusable drugs given intact and upon tampering, preferably in drug abusers and recreational drug users without the medical condition for which the drug is effective.
- the "take again” questionnaire assesses whether subjects would take the abusable drug again if given the opportunity. The patient is asked “If given an opportunity, would you take this drug again? (circle one: YES or NO). This questionnaire can be used to examine the overall desirability of the drug experience with the abusable drugs taken intact and taken after tampering, preferably in drug abusers and recreational drug users without the medical condition for which the drug is effective.
- VAS visual analog scale
- Three performance tasks may be employed for measuring skills related to driving.
- the "critical tracking task” measures the patient's ability to control a displayed error signal in a first-order compensatory tracking task.
- the error is displayed as a horizontal deviation of a cursor from the midpoint on a horizontal, linear scale.
- Compensatory joystick movements correct the error by returning the cursor to the midpoint.
- the frequency at which the patient loses the control is the critical frequency.
- the critical tracking task measures the psychomotor control during a closed loop operation. It is a laboratory analog to on-the-road tracking performance.
- the "stop signal task” measures motor impulsivity, which is defined as the inability to inhibit an activated or pre-cued response leading to errors of commission.
- the task requires patients to make quick key responses to visual go signals, i.e. the letters ABCD presented one at a time in the middle of the screen, and to inhibit any response when a visual stop signal, i.e. "*" in one of the four corners of the screen, is presented at predefined delays.
- the main dependent variable is the stop reaction time on stop signal trials that represents the estimated mean time required to inhibit a response.
- the Tower of London is a decision-making task that measures executive function and planning.
- the task consists of computer generated images of begin- and end-arrangements of three colored balls on three sticks.
- the subject's task is to determine as quickly as possible, whether the end- arrangement can be accomplished by "moving" the balls in two to five steps from the beginning arrangement by pushing the corresponding number coded button.
- the total number of correct decisions is the main performance measure.
- the mean ratio of the time to confirmed perceptible pain relief after administration of the tampered dosage form to the time to confirmed perceptible pain relief after administration of the intact dosage form is more than 20: 1.
- the mean ratio using the aforementioned test method is more than about 15: 1, or more than about 10: 1, or more than about 7: 1, or more than about 5:1, or more than about 3:1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than 1.15: 1.
- the mean ratio of the time to meaningful pain relief after administration of the ' tampered dosage form to the time to meaningful pain relief after administration of the intact dosage form is more than 20:1.
- the mean ratio using the aforementioned test method is more than " about 15:1, or more than about 10: 1, or more than about 7: 1 , or more than about 5: 1 , or more than about 3 : 1 , or more than about 2: 1 , or more than about 1.5:1, or more than about 1.25:1, or more than 1.15: 1.
- the mean ratio of the peak pain intensity difference score after administration of the intact dosage form to the peak pain intensity difference score after administration of the tampered dosage form is more than 10: 1.
- the mean ratio using the aforementioned test method is more than about 8: 1, or more than about 7: 1, or more than about 5: 1, or more than about 3: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1.
- the mean ratio of the peak pain relief score after administration of the intact dosage form to the peak pain relief score after administration of the tampered dosage form is more than 10: 1.
- the mean ratio using the aforementioned test method is more than about 8: 1, or more than about 7: 1, or more than about 5: 1, or more than about 3: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15: 1.
- the mean ratio of change from baseline to two hours post-dose or four hours post-dose in pain intensity score after administration of the intact dosage form to the change from baseline to two hours post-dose in pain intensity score after administration of the tampered dosage form is more than 10: 1; said pain score measured in acute postsurgical pan.
- the mean ratio using the aforementioned test method is more than about 9: 1, or more than about 8: 1, or more than about 7: 1, or more than about 5: 1, or more than about 3:1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1 , or more than about 1.15: 1.
- the mean ratio of the number of patients with pain who obtain 33% pain relief after administration of the intact dosage form when compared with following administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1.
- the mean ratio of the incidence and nausea intensity score in healthy subjects (previously na ⁇ ve to opioid agonist) after administration of the intact dosage form when compared with following administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2:1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 :1.
- the mean ratio of the nausea intensity score in recreational drug users or drug addicts after administration of the tampered dosage form when compared with administration of the intact dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1: 1; said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of moderate or severe sedation or drowsiness in healthy subjects (naive to opioid agonist) after administration of the intact dosage form when compared with following administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15:1, or more than about 1 : 1.
- the aforementioned sedation is measured at 0.5, 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
- the mean ratio of moderate or severe sedation or drowsiness in healthy subjects (na ⁇ ve to opioid agonist) after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 : 1; said dosage form administration followed about 0.5 hour later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of incidence of moderate to severe headache in recreational drug users or drug addicts after administration of the tampered dosage form when compared with administration of the intact dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of the incidence of vomiting or retching in recreational drug users or drug addicts after administration of the tampered dosage form when compared with administration of the intact dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of the incidence of cutaneous flushing in recreational drug users or drug addicts after administration of the tampered dosage form when compared with following administration of the intact dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, said dosage form administration preceded ⁇ by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the sequestered agent is an alcohol deterrent or alcohol aversive agent
- the mean ratio of the incidence of self-reported signs and symptoms selected from the group comprising skin flushing, throbbing, headache, difficulty breathing, nausea, vomiting, retching, sweating, thirst, chest pain, dizziness, fainting, anxiety, uneasiness, weakness, blurred vision and confusion, in recreational drug users or drug addicts after administration of the tampered dosage form when compared with following administration of the intact dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2:1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15:1, said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of the drug liking score in drug abusers and recreational drug users after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1:1.
- the aforementioned drug liking score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
- the mean ratio of the score on the "take again" questionnaire in drug abusers and recreational drug users after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 :1.
- the aforementioned take again score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
- the mean ratio of the score on the "coasting" questionnaire in drug abusers and recreational drug users after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2:1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15: 1, or more than about 1: 1.
- the aforementioned coasting score is measured at 2, 3, 4, 5 or 6 hours, or up to 2, 3, 4, or 6 hours post-dose.
- the mean ratio of impairment on the "critical tracking task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15:1, or more than about 1:1.
- the aforementioned critical tracking task score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
- the mean ratio of impairment on the "critical tracking task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1: 1; said "critical tracking task" driving skills test score measured 2.5 to 6 hours after administration of the dosage form, said dosage form administration followed about 1 hours later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of impairment on the "stop signal task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15:1, or more than about 1 : 1.
- the aforementioned stop signal task score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
- the mean ratio of impairment on the "stop signal task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 : 1; said "critical tracking task" driving skills test score measured 2.5 to 6 hours after administration of the dosage form, said dosage form administration followed about 1 hours later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- the mean ratio of impairment on the "Tower of London" driving skills test score in opioid agonist na ⁇ ve healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1.
- the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3:1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1; or more than about 1 : 1.
- the aforementioned Tower of London score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
- the mean ratio of impairment on the "Tower of London" driving skills test score in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1.
- the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5:1, or more than about 2:1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1:1; said "critical tracking task" driving skills test score measured 2.5 to 6 hours after administration of the dosage form, said dosage form administration followed about 1 hours later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
- composition and methods of the present invention comprise one or more opioid agonists in releasable form and one or more aversive agents in sequestered (i.e., non-releasable or substantially releasable) form chosen from the group comprising cannabinoid antagonists, and alcohol deterrents, and combinations thereof.
- the dosage form optionally comprises, in addition to the foregoing, one of more abuse intervention agents in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form in unsalified form xenobiotics base or pharmaceutically acceptable salts in racemic or enantiomeric form, or mixtures thereof in and they are intended for oral administration.
- All oral pharmaceutical dosage forms of the invention are contemplated, including oral suspensions, tablets, capsules, lozenges, effervescent tablets, effervescent powders, powders, solutions, powders for reconstitution, transmucosal films, buccal products, oral mucoretentive products, oral gastroretentive tablets and capsules, orally disintegrating tablets, fast dissolving tablets, fast dispersing tablets, fast disintegrating dosage forms, administered as immediate release, modified release, enteric coated, sustained release, controlled release, pulsatile release, delayed release, colonic delivery, targeted delivery and extended release dosage form.
- Formulation Technology Emulsions, Suspensions, Solid Forms, Wiley- VCH, 2001; Niazi S and Niazi SK (all of which are hereby incorporated by reference).
- a majority of oral dosage forms commercially available world wide are formulated as immediate release products.
- a wide variety of immediate release dosage forms can be formulated, including oral suspensions, tablets, capsules, lozenges, effervescent tablets, effervescent powders, powders, solutions, powders for reconstitution.
- Aversive Agent e.g., Cannabinoid Antagonist
- the opioid agonist is in an immediate release form and the aversive agent is in a substantially non- releasable form.
- an aversive agent e.g., cannabinoid antagonist
- aversive agent in a substantially non-releasable form may be prepared by combining the aversive agent (e.g., antagonist) with one or more of a pharmaceutically acceptable hydrophobic material.
- aversive agent e.g., cannabinoid antagonist
- particles may be coated with coating that substantially prevents the release of the aversive agent (e..g., antagonist), the coating comprising the hydrophobic materials(s).
- the pharmaceutical acceptable hydrophobic material comprises a cellulose polymer selected from the group consisting of ethylcellulose, cellulose acetate, cellulose propionate (lower, medium or higher molecular weight), cellulose acetate propionate, cellulose acetate butyrate, cellulose acetate phthalate and cellulose triacetate.
- ethylcellulose is one that has an ethoxy content of 44 to 55%. Ethylcellulose may be used in the form of an alcoholic solution.
- the hydrophobic material comprises polylactic acid, polyglycolic acid or a co-polymer of the polylactic and polyglycolic acid.
- the hydrophobic material may comprise a cellulose polymer selected from the group consisting of cellulose ether, cellulose ester, cellulose ester ether, and cellulose.
- the cellulosic polymers have a degree of substitution, D. S., on the anhydroglucose unit, from greater than zero and up to 3 inclusive.
- degree of substitution is meant the average number of hydroxyl groups present on the anhydroglucose unit comprising the cellulose polymer that are replaced by a substituting group.
- Representative materials include a polymer selected from the group consisting of cellulose acylate, cellulose diacylate, cellulose triacylate, cellulose acetate, cellulose diacetate, cellulose triacetate, mono, di, and tricellulose alkanylates, mono, di, and tricellulose aroylates, and mono, di, and tricellulose alkenylates.
- Exemplary polymers include cellulose acetate having a D.S. and an acetyl content up to 21%; cellulose acetate having an acetyl content up to 32 to 39.8%; cellulose acetale having a D.S. of 1 to 2 and an acetyl content of 21 to 35%; cellulose acetate having a D.S. of 2 to 3 and an acetyl content of 35 to 44.8%.
- More specific cellulosic polymers include cellulose propionate having a D.S. of 1.8 and a propyl content of 39.2 to 45 and a hydroxyl content of 2.8 to 5.4%;, cellulose acetate butyrate having a D.S. of 1.8, an acetyl content of 13 to 15% and a butyryl content of 34 to 39%; cellulose acetate butyrate having an acetyl content of 2 to 29%, a butyryl content of 17 to 53% and a hydroxyl content of 0.5 to 4.7%; cellulose triacylate having a D.S.
- cellulose diacylates having a D. S. of 2.2 to 2.6 such as cellulose disuccinate, cellulose dipalmitate, cellulose dioctanoate, cellulose dipentanoate, and coesters of cellulose such as cellulose acetate butyrate, cellulose acetate octanoate butyrate and cellulose acetate propionate.
- a substantially non-releasable form in a substantially non-releasable form includes acetaldehyde dimethyl cellulose acetate, cellulose acetate ethylcarbamate, cellulose acetate methylcarbamate, and cellulose acetate dimethylaminocellulose acetate.
- An acrylic polymer useful for preparation of the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form includes, but are not limited to, acrylic resins comprising copolymers synthesized from acrylic and methacrylic acid esters (e.g., the copolymer of acrylic acid lower alkyl ester and methacrylic acid lower alkyl ester) containing about 0.02 to 0.03 mole of a tri (lower alkyl) ammonium group per mole of the acrylic and methacrylic monomers used.
- An example of a suitable acrylic resin is Eudragit RS. Eudragit RS30D is preferred.
- Eudragit RS is a water insoluble copolymer of ethyl acrylate (EA), methyl methacrylate (MM) and trimethylammoniumethyl methacrylate chloride (TAM) in which the molar ratio of TAM to the remaining components (EA and MM) is 1 :40.
- Acrylic resins such as Eudragit RS may be used in the form of an aqueous suspension.
- the acrylic polymer may be selected from the group consisting of acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamide copolymer, poly(methyl methacrylate), polymethacrylate, poly(methyl methacrylate) copolymer, polyacrylamide, aminoalkyl methacrylate copolymer, poly(methacrylic acid anhydride), and glycidyl methacrylate co-polymers.
- aversive agent e.g., cannabinoid antagonist
- suitable plasticizers e.g., acetyl triethyl citrate and/or acetyl tributyl citrate may also be admixed with the polymer.
- the coating may also contain additives such as coloring agents, talc and/or magnesium stearate, which are well known in the coating art.
- the coating composition may be applied onto the aversive agent (e.g., cannabinoid antagonist) particles by spraying it onto the particles using any suitable spray equipment known in the part.
- aversive agent e.g., cannabinoid antagonist
- a Wuster fluidized- bed system may be used in which an air jet, injected from underneath, fluidizes the coated material and effects drying while the insoluble polymer coating is sprayed on.
- the thickness of the coating will depend on the characteristics of the particular coating composition being used. However, it is well within the ability of one skilled in the art to determine by routine experimentation the optimum thickness of a particular coating required for a particular dosage form of the present invention.
- the pharmaceutically acceptable hydrophobic material useful for preparing a cannabinoid antagonist in a substantially non-releasable form includes a biodegradable polymer comprising a poly(lactic/glycolic acid) ("PLGA"), a polylactide, a polyglycolide, a polyanhydride, a polyorthoester, polycaprolactones, polyphosphazenes, polysaccharides, proteinaceous polymers, polyesthers, polydioxanone, polygluconate, polylactic-acid- polyethylene oxide copolymers, poly(hydroxybutyrate), polyphosphoesther or mixtures or blends of any of these.
- PLGA poly(lactic/glycolic acid)
- biodegradable polymer comprises a poly(lactic/glycolic acid), a copolymer of lactic and glycolic acid, having molecular weight of about 2,000 to about 500,000 daltons.
- the ratio of lactic acid to glycolic acid is from about 100:0 to about 25:75, with the ratio of lactic acid to glycolic acid of 65:35 being preferred.
- Poly(lactic/glycolic acid) may be prepared by the procedure set forth in
- Ludwig prepares the copolymer by condensation of lactic acid and glycolic acid in the presence of a readily removable polymerization catalyst (e.g., a strong acid ion-exchange resin such as Dowex HCR-W2-H).
- a readily removable polymerization catalyst e.g., a strong acid ion-exchange resin such as Dowex HCR-W2-H.
- the amount of catalyst is not critical to the polymerization, but typically is from about 0.01 to about 20 parts by weight relative to the total weight of combined lactic acid and glycolic acid.
- the polymerization reaction may be conducted without solvents at a temperature from about 100 C. to about 250 C.
- Poly(lactic/glycolic acid) is then recovered by filtering the molten reaction mixture in an organic solvent such as dichloromethane or acetone and then filtering to remove the catalyst.
- the aversive agent e.g., cannabinoid antagonist
- an opioid agonist e.g., cannabinoid antagonist
- the oral dosage form is a capsule or a tablet.
- the aversive agent e.g., cannabinoid antagonist
- the aversive agent and agonist may be combined with one or more inert, non-toxic pharmaceutical excipients which are suitable for the manufacture of tablets.
- excipients include, for example, an inert diluent such as lactose; granulating and disintegrating agents such as cornstarch; binding agents such as starch; and lubricating agents such as magnesium stearate.
- the oral dosage form of the present invention may be formulated to provide immediate release of the opioid agonist contained therein. In other embodiments of the invention, however, the oral dosage form provides sustained-release of the opioid agonist.
- the oral dosage forms providing sustained release of the opioid agonist may be prepared by admixing the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form with the agonist and desirable pharmaceutical excipients to provide a tablet, and then coating the tablet with a sustained-release tablet coating.
- aversive agent e.g., cannabinoid antagonist
- sustained release opioid agonist tablets may be prepared by admixing the substantially non-releasable form of an aversive agent (e.g., cannabinoid antagonist) with an aversive agent (e.g., cannabinoid antagonist) in a matrix that provides the tablets with sustained-releasing properties.
- an aversive agent e.g., cannabinoid antagonist
- an aversive agent e.g., cannabinoid antagonist
- the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 6 hours comprising (i) an aversive agent in releasable form and (ii) a substantially non-releasable aversive agent.
- the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 8 hours comprising (i) an opioid agonist in releasable form and (ii) a substantially non-releasable aversive agent.
- the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 12 hours comprising (i) an opioid agonist in releasable form and (ii) a substantially non-releasable aversive agent.
- the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 24 hours comprising (i) an opioid agonist in releasable form and (ii) a substantially non-releasable aversive agent.
- a combination of the opioid agonist and a substantially non-releasable form of an aversive agent may be formulated as a controlled or sustained release oral formulation in any suitable tablet, coated tablet or multiparticulate formulation known to those skilled in the art.
- the sustained release dosage form may optionally include a sustained release carrier which is incorporated into a matrix along with the opioid agonist and a non-available form of an aversive agent (e.g., cannabinoid antagonist), or may be applied as a sustained release coating.
- the sustained release dosage form comprises such particles comprising the opioid agonist, wherein the particles have diameter from about 0.1 mm to about 2.5 mm, preferably from about 0.5 mm to about 2 mm.
- the opioid agonist particles are preferably film coated with a material that permits release of the opioid agonist at a sustained rate in an aqueous medium.
- the film coat is chosen so as to achieve, in combination with the other stated properties, a desired in-vitro release rate.
- the sustained release coating formulations of the present invention should be capable of producing a strong, continuous film that is smooth and elegant, capable of supporting pigments and other coating additives, non-toxic and inert.
- the dosage forms comprising an opioid agonist and a substantially non-releasable aversive agent may optionally be coated with one or more materials suitable for the modulation of the opioid agonist release or for the protection of the formulation.
- coatings are provided to permit either pH-dependent or pH-independent release, e.g., when exposed to gastrointestinal fluid.
- a pH-dependent coating serves to release the cannabinoid in desired areas of the gastro-intestinal (GI) tract, e.g., the stomach or small intestine, such that an absorption profile is provided which is capable of providing at least about six hours and preferably about twelve hours to up to about twenty-four hours of therapeutic effect to a patient.
- the coating is designed to achieve optimal release of the cannabinoid regardless of pH-changes in the environmental fluid, e.g., the GI tract. It is also possible to formulate compositions which release a portion of the dose in one desired area of the GI tract, e.g., the stomach, and release the remainder of the dose in another area of the GI tract, e.g., the small intestine. [00335] Formulations according to the invention that utilize pH-dependent coatings to obtain formulations may also impart a repeat-action effect whereby unprotected drug is coated over the enteric coat and is released in the stomach, while the remainder, being protected by the enteric coating,-is released further down the gastrointestinal tract.
- Coatings which are pH-dependent may be used in accordance with the present invention include shellac, cellulose acetate phthalate (CAP), polyvinyl acetate phthalate (PVAP), hydroxypropyl methylcellulose phthalate, and methacrylic acid ester copolymers, zein, and the like.
- CAP cellulose acetate phthalate
- PVAP polyvinyl acetate phthalate
- zein methacrylic acid ester copolymers
- the substrate e.g., tablet core bead, matrix particle
- the cannabinoid analgesic is coated with a hydrophobic material selected from (i) an alkylcellulose; (ii) an acrylic polymer; or (iii) mixtures thereof.
- the coating may be applied in the form of an organic or aqueous solution or dispersion. The coating may be applied to obtain a weight gain from about 2 to about 25% of the substrate in order to obtain a desired sustained release profile.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists, and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
- the dosage form provides said an in-vitro release rate of from 0% to about 40% at 1 hour, from about 5% to about 55% at 2 hours, from about 10% to about 60% at 4 hours, from about 15% to about 70% at 6 hours, from about 25% to about 80% at 9 hours, and greater than about 50% at 12 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours; said in-vitro release rate being substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist or pharmaceutically acceptable salts thereof or mixture
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for once-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of from 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for once-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of from 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours; said in-vitro release rate being substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and
- the opioid agonist dosage forms provide an in-vitro release of from 2% to about 50% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0 C.
- SGF Simulated Gastric Fluid
- the opioid agonist dosage form provides an in-vitro release from about 5% to about 45% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0 C.
- SGF Simulated Gastric Fluid
- the opioid agonist dosage form provides a C max of opioid agonist which is less than 65% of the C m a x of an equivalent dose of an oral immediate release opioid agonist solution.
- the dosage form provides a time to
- the dosage from maintains a plasman opioid agonist concentration within 50% of C max for about 1 to about 9 hours during the 12 hour dosing interval.
- the dosage from maintains a plasman opioid agonist concentration within 50% of C max for about 1 to about 9 hours during the 24 hour dosing interval.
- the dosage form provides a T max of opioid agonist at a time point 1.5 to 20 times later than the T ma ⁇ provided by an equivalent dose of an immediate release opioid agonist solution.
- a T max at a time point about 1.5 to 15 times late, or about of 1.5 to 10 times later, or about of 1.5 to 7 times later, or about of 1.5 to 3 times later, or about of 2 to 20 times later, or about of 2 to 10 times later, or about of 2 to 5 times later, or about of 2 to 3 times later, or about of 2.5 to 20 times later, or about of 2.5 to 8 times later, or about of 2.5 to 5 times later, or about of 2.5 to 4 times later, or about of 3 to 20 times later, or about of 3 to 10 times later, or about of 3 to 5 times later.
- the dosage form provides a mean in vivo extent of absorption of opioid agonist from 0 to 4 hours which is at least 20% of the mean in vivo extent of absorption from to 0 to 12 hours, wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve from the time of drug administration to the specified time point.
- said in vivo extent of absorption from 0 to 4 hours is at least about 5%, or at least about 10%, or at least about 15%, or at least about 25%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, at least about 90%, or about 100% of the mean in vivo extent of absorption from to 0 to 12 hours.
- the dosage form provides a mean in vivo extent of absorption of opioid agonist from 0 to 8 hours which is at least 20% of the mean in vivo extent of absorption from to 0 to 24 hours, wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve from the time of drug administration to the specified time point.
- said in vivo extent of absorption from 0 to 8 hours is at least about 5%, or at least about 10%, or at least about 15%, or at least about 25%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, at least about 90%, or about 100% of the mean in vivo extent of absorption from to 0 to 24 hours.
- the dosage form provides a mean in vivo extent of absorption of opioid agonist from 0 to 12 hours which is at least 20% of the mean in vivo extent of absorption from to 0 to 24 hours, wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve from the time of drug administration to the specified time point.
- said in vivo extent of absorption from 0 to 12 hours is at least about 5%, or at least about 10%, or at least about 15%, or at least about 25%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, at least about 90%, or about 100% of the mean in vivo extent of absorption from to 0 to 24 hours.
- the dosage form provides a mean in vivo extent of absorption of opioid agonist over the dosing interval (e.g., from 0 to 12 hours or from 0 to 24 hours) which is at least 40% of the mean in vivo extent of absorption from to 0 to ⁇ , wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve (AUC) from the time of drug administration to the specified time point and where AUC infinity is the sum of AUC from time "0" to time "t" (the last quantifiable time point which has been sampled) plus the extrapolated AUC from the last quantifiable sampling time point to infinity.
- AUC concentration time curve
- the dosage form provides an oral pharmaceutical composition of opioid agonist or a pharmaceutically acceptable salt thereof or mixtures thereof, said dosage form providing an in- vitro release of between 0% to about 50% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0 C.
- SGF Simulated Gastric Fluid
- the dosage form provides an oral pharmaceutical composition of opioid agonist or a pharmaceutically acceptable salt thereof or mixtures thereof, said dosage form providing an in- vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% to about 80% at 0.5 hours, and greater than about 40% at 1 hour; or between 0% to about 90% at 0.5 hours, and greater than about 60% at 1 hour; or between 1.6 and 7.2 at 37 0 C of between 0% to about 100% at 0.5 hours, and greater than about 60% at 1 hour.
- the dosage form provides an oral pharmaceutical composition of opioid agonist or a pharmaceutically acceptable salt thereof or mixtures thereof, said dosage form providing an in- vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% to about 90% at 1 hour, and greater than about 40% at 2 hours; or between 1.6 and 7.2 at 37 0 C of between 0% to about 90% at 1 hour, and greater than about 70% at 2 hours; or between 1.6 and 7.2 at 37 0 C of between 0% to about 50% at 1 hour, and greater than about 30% at 2 hours; between 1.6 and 7.2 at 37 0 C of between 0% to about 30% at 1 hour, and greater than about 25% at 2 hours; or between 1.6 and 7.2 at 37 0 C of between 0% to about 100% at 1 hour, and greater than about 60% at 2 hours.
- the invention comprises an oral pharmaceutical dosage form providing an in-vitro release of between 0% to about 50% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0 C.
- SGF Simulated Gastric Fluid
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 3% and about 95% at 4 hours and between about 10% and about 100% at 8 hours; or between 10% and about 65% at 1 hour, between about 20% and about 75% at 2 hours, between about 30% and about 95% at 4 hours and between about 40% and about 100% at 8 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 5% and about 50% at 1 hour, between about 10% and about 75% at 2 hours, between about 20% and about 95% at 4 hours, between about 40% and about 100% at 8 hours, greater than about 50% at 12 hours, greater than about 70% at 18 hours, and greater than about 80% at 24 hours; or between 5% and about 50% at 1 hour, between about 10% and about 75% at 2 hours, between about 20% and about 95% at 4 hours, between
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 20% and about 50% at 1 hour, between about 40% and about 75% at 2 hours, between about 60% and about 95% at 4 hours, between about 80% and about 100% at 8 hours and between about 90% and about 100% at 12 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 10% and about 95% at 4 hours, between about 35% and about 100% at 8 hours, between about 55% and about 100% at 12 hours, between about 70% to about 100% at 16 hours, and greater than about 90% at 24 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 30% at 1 hour, between about 0% and about 45% at 2 hours, between about 3% and about 55% at 4 hours, between about 10% and about 65% at 8 hours, between about 20% and about 75% at 12 hours, between about 30% to about 88% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 20% and about 100% at 12 hours, between about 30% to about 100% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 15% and about 25% at 1 hour, between about 25% and about 35% at 2 hours, between about 30% and about 45% at 4 hours, between about 40% and about 60% at 8 hours, between about 55% and about 70% at 12 hours and between about 60% to about 75% at 16 hours.
- the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist which is substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0 C of between 0% to about 50% by weight of the opioid agonist.
- SGF Simulated Gastric Fluid
- said release rate is between 0% to about 1%, or 0% to about 3%, or 0% to about 5%, or 0% to about 10%, or 0% to about 15%, or 0% to about 20%, 0% to about 30%, or 0% to about 40%, or 0% to about 60%, or 0% to about 70%, or 0% to about 80%, or 0% to about 90%, 0% to about 100%.
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 3% and about 95% at 4 hours and between about 10% and about 100% at 8 hours.
- said release rate is between 0% and about 10% at 1 hour, between about 0% and about 20% at 2 hours, between about 2% and about 80% at 4 hours and between about 5% and about 100% at 8 hours; or between 0% and about 20% at 1 hour, between about 0% and about 40% at 2 hours, between about 0% and about 80% at 4 hours and between about 2% and about 100% at 8 hours; or between 0% and about 40% at 1 hour, between about 0% and about 60% at 2 hours, between about 5% and about 85% at 4 hours and between about 5% and about 90% at 8 hours and greater than 20% at 12 hours; or between 0% and about 50% at 1 hour, between about 0% and about 50% at 2 hours, between about 10% and about 90% at 4 hours and between about 15% and about 90% at 8 hours and greater than 30% at 12 hours; or between 0% and about 70% at 1 hour, between about 0% and about 70% at 2 hours, between about 10% and about 75% at 4 hours and between about 15% and about 90% at 8 hours and greater than 30% at 12 hours.
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 10% and about 65% at 1 hour, between about 20% and about 75% at 2 hours, between about 30% and about 95% at 4 hours and between about 40% and about 100% at 8 hours.
- said release rate is between 2% and about 70% at 1 hour, between about 5% and about 80% at 2 hours, between about 10% and about 90% at 4 hours and between about 20% and about 100% at 8 hours; or between 5% and about 60% at 1 hour, between about 10% and about 75% at 2 hours, between about 15% and about 85% at 4 hours and between about 30% and about 100% at 8 hours; or between 20% and about 70% at 1 hour, between about 20% and about 75% at 2 hours, between about 20% and about 90% at 4 hours and between about 40% and about 100% at 8 hours; or between 30% and about 80% at 1 hour, between about 40% and about 85% at 2 hours, between about 40% and about 90% at 4 hours and between about 60% and about 100% at 8 hours; or between 1% and about 20% at 1 hour, between about 5% and about 20% at 2 hours, between about 10% and about 40% at 4 hours and between about 20% and about 40% at 8 hours and greater than 40% at 12 hours.
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
- said release rate is between 0% to about 30% at 1 hour, from about 5% to about 45% at 2 hours, from about 10% to about 60% at 4 hours, from about 15% to about 70% at 6 hours, from about 25% to about 80% at 9 hours, and greater than about 50% at 12 hours; or between 0% to about 20% at 1 hour, from about 2% to about 35% at 2 hours, from about 5% to about 50% at 4 hours, from about 10% to about 60% at 6 hours, from about 15% to about 70% at 9 hours, and greater than about 40% at 12 hours; or between 0% to about 10% at 1 hour, from about 1% to about 30% at 2 hours, from about 5% to about 40% at 4 hours, from about 10% to about 60% at 6 hours, from about 15% to about 70% at 9 hours, and greater than about 40% at 12 hours; or between 0% to about 5% at 1 hour, from about 0% to about 10% at 2 hours, from about 2% to about 20% at 4 hours, from about 5% to about 30% at 6 hours, from about 10% to about 40% at 9 hours, and greater than about 50% at 12 hours
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 5% and about 50% at 1 hour, between about 10% and about 75% at 2 hours, between about 20% and about 95% at 4 hours, between about 40% and about 100% at 8 hours, greater than about 50% at 12 hours, greater than about 70% at 18 hours, and greater than about 80% at 24 hours.
- said release rate is between 2% and about 50% at 1 hour, between about 5% and about 75% at 2 hours, between about 15% and about 75% at 4 hours, between about 30% and about 90% at 8 hours, greater than about 40% at 12 hours, greater than about 60% at 18 hours, and greater than about 70% at 24 hours; or between 1% and about 40% at 1 hour, between about 2% and about 60% at 2 hours, between about 10% and about 65% at 4 hours, between about 20% and about 80% at 8 hours, greater than about 30% at 12 hours, greater than about 40% at 18 hours, and greater than about 60% at 24 hours; or between 5% and about 60% at 1 hour, between about 15% and about 80% at 2 hours, between about 25% and about 95% at 4 hours, between about 45% and about 100% at 8 hours, greater than about 60% at 12 hours, greater than about 80% at 18 hours, and greater than about 90% at 24 hours; or between 10% and about 65% at 1 hour, between about 20% and about 85% at 2 hours, between about 30% and about 100% at 4 hours, between about 60% and about 100% at 4 hours, between
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours.
- said release rate is between 0% to about 20% at 1 hour, from about 5% to about 50% at 4 hours, from about 10% to about 60% at 8 hours, from about 15% to about 70% at 12 hours, from about 25% to about 90% at 18 hours, and greater than about 55% at 24 hours; or between 0% to about 10% at 1 hour, from about 5% to about 40% at 4 hours, from about 8% to about 50% at 8 hours, from about 10% to about 60% at 12 hours, from about 22% to about 80% at 18 hours, and greater than about 45% at 24 hours; or between 0% to about 35% at 1 hour, from about 15% to about 70% at 4 hours, from about 25% to about 75% at 8 hours, from about 30% to about 85% at 12 hours, from about 40% to about 100% at 18 hours, and greater than about 75% at 24 hours; or between 0% to about 40% at 1 hour, from about 20% to about 70% at 4 hours, from about 30% to about 80% at 8 hours, from about 35% to about 90% at 12 hours, from about 45% to about 100% at 18 hours, and
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in- vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 25% and about 100% at 12 hours, between about 30% and about 100% at 16 hours, between about 50% and about 100% at 24 hours, and greater than about 80% at 36 hours.
- said release rate is between 0% and about 40% at 1 hour, between about 0% and about 65% at 2 hours, between about 2% and about 85% at 4 hours, between about 8% and about 90% at 8 hours, between about 20% and about 95% at 12 hours, between about 25% and about 95% at 16 hours, between about 40% and about 90% at 24 hours, and greater than about 70% at 36 hours; or between 0% and about 30% at 1 hour, between about 0% and about 50% at 2 hours, between about 1% and about 75% at 4 hours, between about 5% and about 80% at 8 hours, between about 10% and about 85% at 12 hours, between about 15% and about 90% at 16 hours, between about 30% and about 80% at 24 hours, and greater than about 70% at 36 hours; or between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 5% and about 100% at 4 hours, between about 15% and about 100% at 8 hours, between about 35% and about 100% at 12 hours, between about 40% and about 100% at 16 hours, between about 60% and about 100% at
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 20% and about 50% at 1 hour, between about 40% and about 75% at 2 hours, between about 60% and about 95% at 4 hours, between about 80% and about 100% at 8 hours and between about 90% and about 100% at 12 hours.
- said release rate is between 15% and about 45% at 1 hour, between about 35% and about 70% at 2 hours, between about 55% and about 90% at 4 hours, between about 75% and about 90% at 8 hours and between about 80% and about 95% at 12 hours; or between 10% and about 40% at 1 hour, between about 30% and about 65% at 2 hours, between about 50% and about 85% at 4 hours, between about 70% and about 85% at 8 hours and between about 75% and about 90% at 12 hours; or between 5% and about 35% at 1 hour, between about 25% and about 60% at 2 hours, between about 45% and about 80% at 4 hours, between about 65% and about 80% at 8 hours and between about 70% and about 85% at 12 hours; or between 25% and about 55% at 1 hour, between about 45% and about 80% at 2 hours, between about 65% and about 95% at 4 hours, between about 85% and about 100% at 8 hours and between about 95% and about 100% at 12 hours; or between 30% and about 60% at 1 hour, between about 50% and about 80% at 2 hours, between 30% and about 60% at
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 10% and about 95% at 4 hours, between about 35% and about 100% at 8 hours, between about 55% and about 100% at 12 hours, between about 70% to about 100% at 16 hours, and greater than about 90% at 24 hours.
- said release rate is between 0% and about 40% at 1 hour, between about 0% and about 65% at 2 hours, between about 8% and about 85% at 4 hours, between about 30% and about 90% at 8 hours, between about 45% and about 100% at 12 hours, between about 60% to about 100% at 16 hours, and greater than about 80% at 24 hours; or between 0% and about 30% at 1 hour, between about 0% and about 55% at 2 hours, between about 5% and about 75% at 4 hours, between about 20% and about 80% at 8 hours, between about 35% and about 100% at 12 hours, between about 50% to about 100% at 16 hours, and greater than about 70% at 24 hours; or between 0% and about 20% at 1 hour, between about 0% and about 45% at 2 hours, between about 5% and about 65% at 4 hours, between about 10% and about 70% at 8 hours, between about 25% and about 80% at 12 hours, between about 40% to about 100% at 16 hours, and greater than about 60% at 24 hours; or between 0% and about 60% at 1 hour, between about 0% and about 80%
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 30% at 1 hour, between about 0% and about 45% at 2 hours, between about 3% and about 55% at 4 hours, between about 10% and about 65% at 8 hours, between about 20% and about 75% at 12 hours, between about 30% to about 88% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours.
- said release rate is between 0% and about 25% at 1 hour, between about 0% and about 40% at 2 hours, between about 2% and about 50% at 4 hours, between about 8% and about 60% at 8 hours, between about 10% and about 70% at 12 hours, between about 25% to about 80% at 16 hours, between about 45% and about 100% hours at 24 hours and greater than 75% at 36 hours; or between 0% and about 20% at 1 hour, between about 0% and about 35% at 2 hours, between about 1% and about 45% at 4 hours, between about 5% and about 55% at 8 hours, between about 8% and about 65% at 12 hours, between about 20% to about 75% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 15% at 1 hour, between about 0% and about 30% at 2 hours, between about 0% and about 40% at 4 hours, between about 5% and about 50% at 8 hours, between about 8% and about 60% at 12 hours, between about 15% to about 70% at 16 hours, between about 35% and about 100% hours
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 20% and about 100% at 12 hours, between about 30% to about 100% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours.
- said release rate is between 0% and about 45% at 1 hour, between about 0% and about 70% at 2 hours, between about 3% and about 90% at 4 hours, between about 8% and about 100% at 8 hours, between about 15% and about 100% at 12 hours, between about 25% to about 100% at 16 hours, between about 45% and about 100% hours at 24 hours and greater than 80% at 36 hours; or between 0% and about 40% at 1 hour, between about 0% and about 65% at 2 hours, between about 0% and about 80% at 4 hours, between about 5% and about 80% at 8 hours, between about 10% and about 90% at 12 hours, between about 20% to about 100% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 35% at 1 hour, between about 0% and about 60% at 2 hours, between about 0% and about 70% at 4 hours, between about 3% and about 70% at 8 hours, between about 5% and about 80% at 12 hours, between about 15% to about 100% at 16 hours, between about 30% and about 100% hours at 24
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 15% and about 25% at 1 hour, between about 25% and about 35% at 2 hours, between about 30% and about 45% at 4 hours, between about 40% and about 60% at 8 hours, between about 55% and about 70% at 12 hours and between about 60% to about 75% at 16 hours.
- said release rate is between 10% and about 20% at 1 hour, between about 20% and about 30% at 2 hours, between about 25% and about 40% at 4 hours, between about 30% and about 50% at 8 hours, between about 50% and about 65% at 12 hours and between about 55% to about 65% at 16 hours; or between 5% and about 15% at 1 hour, between about 15% and about 25% at 2 hours, between about 20% and about 35% at 4 hours, between about 25% and about 45% at 8 hours, between about 45% and about 60% at 12 hours and between about 50% to about 60% at 16 hours; or between 15% and about 30% at 1 hour, between about 20% and about 40% at 2 hours, between about 20% and about 50% at 4 hours, between about 30% and about 70% at 8 hours, between about 60% and about 80% at 12 hours and between about 70% to about 90% at 16 hours; or between 0% and about 50% at 1 hour, between about 5% and about 50% at 2 hours, between about 5% and about 70% at 4 hours, between about 10% and about 80% at 8 hours, between about 20% and about 100% at 12 hours and between about 40% to
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said in-vitro release rate being substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
- the difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH using the aforementioned methods is no greater than 50%, or no greater than 4,0%, or no greater than 35%, or no greater than 25%, or no greater than 20%, or no greater than 15%, or no greater than 10%, or no greater than 5%.
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said dosage forms of opioid agonist providing in-vitro release rates by weight of between 0% to about 50% by weight of the opioid agonist from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0 C.
- SGF Simulated Gastric Fluid
- said release rate at one hour is between 0% to about 10% by weight, or 0% to about 20% by weight, or is between 0% to about 30% by weight, or 0% to about 40% by weight, or between 0% to about 60% by weight, or 0% to about 70% by weight, or 0% to about 80% by weight, or 0% to about 90% by weight, or 10% to about 50% by weight, or 10% to about 60% by weight, or 10% to about 70% by weight, or 10% to about 90% by weight, or 10% to about 100% by weight, or 30% to about 100% by weight, or 50% to about 100% by weight.
- the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said dosage forms of opioid agonist providing in-vitro release rates by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0 C of between 0% to about 80% at 0.5 hours, and greater than about 40% at 1 hour.
- said release rate is between 0% to about 40% at 0.5 hours, and greater than about 60% at 1 hour; or between 0% to about 20% at 0.5 hours, and greater than about 40% at 1 hour; or between 0% to about 20% at 0.5 hours, and greater than about 20% at 1 hour; or between 0% to about 90% at 0.5 hours, and greater than about 60% at 1 hour; or between 0% to about 100% at 0.5 hours, and greater than about 60% at 1 hour; or between 0% to about 90% at 1 hour, and greater than about 40% at 2 hours; or between 0% to about 100% at 1 hour, and greater than about 60% at 2 hours; or between 0% to about 60% at 1 hour, and greater than about 40% at 2 hours; or between 0% to about 40% at 1 hour, and greater than about 30% at 2 hours; or between 0% to about 50% at 1 hour, and greater than about 40% at 2 hours; or between 0% to about 30% at 1 hour, and greater than about 20% at 2 hours; or between 0% and about 50% at 1 hour, between about 0% and about 80% at 2
- some or all of the dissolution embodiments and specifications (e.g., USP Basket Method, USP Paddle Method) of the invention applicable to the opioid agonist of the oral dosage are also applicable to the substantially non-releasable or non-releasable aversive agent (e.g., cannabinoid antagonist or alcohol deterrent) of the dosage form.
- aversive agent e.g., cannabinoid antagonist or alcohol deterrent
- some or all of the pharmacokinetic embodiments and specifications (e.g., C max , T max , AUC, percent absorption) of the invention applicable to the opioid agonist of the oral dosage are also applicable to the substantially non-releasable or non-releasable aversive agent (e.g., cannabinoid antagonist or alcohol deterrent) of the dosage form.
- aversive agent e.g., cannabinoid antagonist or alcohol deterrent
- Cellulosic materials and polymers including alkylcelluloses, provide hydrophobic materials well suited for coating the beads according to the invention.
- one preferred alkylcellulosic polymer is ethylcellulose, although the artisan will appreciate that other cellulose and/or alkylcellulose polymers may be readily employed, singly or in any combination, as all or part of a hydrophobic coating according to the invention.
- Aquacoat is prepared by dissolving the ethylcellulose in a water- immiscible organic solvent and then emulsifying the same in water in the presence of a surfactant and a stabilizer. After homogenization to generate submicron droplets, the organic solvent is evaporated under vacuum to form a pseudolatex.
- the plasticizer is not incorporated in the pseudolatex during the manufacturing phase. Thus, prior to using the same as a coating, it is necessary to intimately mix the Aquacoat with a suitable plasticizer prior to use.
- aqueous dispersion of ethylcellulose is commercially available as Surelease.
- This product is prepared by incorporating plasticizer into the dispersion during the manufacturing process.
- a hot melt of a polymer, plasticizer (dibutyl sebacate), and stabilizer (oleic acid) is prepared as a homogeneous mixture, which is then diluted with an alkaline solution to obtain an aqueous dispersion which can be applied directly onto substrates.
- the hydrophobic material comprising the controlled release coating is a pharmaceutically acceptable acrylic polymer, including but not limited to acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamide copolymer, poly(methyl methacrylate), polymethacrylate, poly(methyl methacrylate) copolymer, polyacrylamide, aminoalkyl methacrylate copolymer, poly(methacrylic acid anhydride), and glycidyl methacrylate copolymers.
- acrylic acid and methacrylic acid copolymers including but not limited to acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, poly(acrylic acid), poly(methacrylic
- the acrylic polymer is comprised of one or more ammonio methacrylate copolymers.
- Ammonio methacrylate copolymers are well known in the art, and are described in NF XVII as fully polymerized copolymers of acrylic and methacrylic acid esters with a low content of quaternary ammonium groups.
- Certain methacrylic acid ester-type polymers are useful for preparing pH-dependent coatings which may be used in accordance with the present invention.
- Eudragit E is an example of a methacrylic acid copolymer which swells and dissolves in acidic media.
- Eudragit L is a methacrylic acid copolymer which does not swell at about pH ⁇ 5.7 and is soluble at about pH>6.
- Eudragit S does not swell at about pH ⁇ 6.5 and is soluble at about pH>7.
- Eudragit RL and Eudragit RS are water swellable, and the amount of water absorbed by these polymers is pH-dependent, however, dosage forms coated with Eudragit RL and RS are pH-independent.
- the acrylic coating comprises a mixture of two acrylic resin lacquers commercially available from under the trade names Eudragit RL30D and Eudragit RS30D, respectively.
- Eudragit RL30D and Eudragit RS30D are copolymers of acrylic and methacrylic esters with a low content of quaternary ammonium groups, the molar ratio of ammonium groups to the remaining neutral (meth)acrylic esters being 1:20 in Eudragit RL30D and 1 :40 in Eudragit RS30D.
- the mean molecular weight is about 150,000.
- the code designations RL (high permeability) and RS (low permeability) refer to the permeability properties of these agents.
- Eudragit RL/RS mixtures are insoluble in water and in digestive fluids. However, coatings formed from the same are swellable and permeable in aqueous solutions and digestive fluids.
- the Eudragit RL/RS dispersions of the present invention may be mixed together in any desired ratio in order to ultimately obtain a sustained release formulation having a desirable dissolution profile. Desirable sustained release formulations may be obtained, for instance, from a retardant coating derived from 100% Eudragit RL, 50% Eudragit RL and 50% Eudragit RS, and 10% Eudragit RL:90%Eudragit RS.
- a retardant coating derived from 100% Eudragit RL, 50% Eudragit RL and 50% Eudragit RS, and 10% Eudragit RL:90%Eudragit RS.
- acrylic polymers may also be used, such as, for example, Eudragit L.
- the inclusion of an effective amount of a plasticizer in the aqueous dispersion of hydrophobic material will further improve the physical properties of the sustained release coating.
- a plasticizer into an ethylcellulose coating containing sustained release coating before using the same as a coating material.
- the amount of plasticizer included in a coating solution is based on the concentration of the film-former, e.g., most often from about 1 to about 50 percent by weight of the film-former. Concentration of the plasticizer, however, can only be properly determined after careful experimentation with the particular coating solution and method of application.
- plasticizers for ethylcellulose include water insoluble plasticizers such as dibutyl sebacate, diethyl phthalate, triethyl citrate, tributyl citrate, and triacetin, although it is possible that other water- insoluble plasticizers (such as acetylated monoglycerides, phthalate esters, castor oil, etc.) may be used.
- Triethyl citrate is an especially preferred plasticizer for the aqueous dispersions of ethyl cellulose of the present invention.
- plasticizers for the acrylic polymers of the present invention include, but are not limited to citric acid esters such as triethyl citrate NF XVI, tributyl citrate, dibutyl phthalate, and possibly 1,2- propylene glycol.
- Other plasticizers which have proved to be suitable for enhancing the elasticity of the films formed from acrylic films such as Eudragit RL/RS lacquer solutions include polyethylene glycols, propylene glycol, diethyl phthalate, castor oil, and triacetin.
- Triethyl citrate is an especially preferred plasticizer for the aqueous dispersions of ethyl cellulose of the present invention.
- a hydrophobic controlled release coating material is used to coat inert pharmaceutical beads such as nu pariel 18/20 beads, which are already coated with an opioid agonist
- a plurality of the resultant solid controlled release beads may thereafter be placed in a gelatin capsule, with the cannabinoid antagonist in a substantially non-releasable form.
- the dosage form provides an effective controlled release dose of the opioid agonist when ingested and contacted by an environmental fluid, e.g., gastric fluid or dissolution media.
- the controlled release bead formulations of the present invention slowly release the opioid agonist, e.g., when ingested and exposed to gastric fluids, and then to intestinal fluids.
- the controlled release profile of the formulations of the invention can be altered, for example, by varying the amount of overcoating with the hydrophobic material, altering the manner in which the plasticizer is added to the hydrophobic material, by varying the amount of plasticizer relative to hydrophobic material, by the inclusion of additional ingredients or excipients, by altering the method of manufacture, etc.
- the dissolution profile of the ultimate product may also be modified, for example, by increasing or decreasing the thickness of the retardant coating.
- Spheroids or beads coated with an opioid agonist may be prepared, e.g., by dissolving the drug in water and then spraying the solution onto a substrate, for example, nu pariel 18/20 beads, using a Wuster insert.
- additional ingredients are also added prior to coating the beads in order to assist the binding of the cannabinoid to the beads, and/or to color the solution, etc.
- a product which includes hydroxypropyl methylcellulose, etc. with or without colorant e.g., Opadry
- the resultant coated substrate in this example beads, may then be optionally overcoated with a barrier agent, to separate the therapeutically active agent from the hydrophobic controlled release coating.
- a barrier agent is one which comprises hydroxypropyl methylcellulose.
- any film-former known in the art may be used. It is preferred that the barrier agent does not affect the dissolution rate of the final product.
- the beads may then be overcoated with an aqueous dispersion of the hydrophobic material.
- the aqueous dispersion of hydrophobic material preferably further includes an effective amount of plasticizer, e.g. triethyl citrate.
- plasticizer e.g. triethyl citrate.
- Pre-formulated aqueous dispersions of ethylcellulose, such as Aquacoat or Surelease, may be used. If Surelease is used, it is not necessary to separately add a plasticizer.
- pre-formulated aqueous dispersions of acrylic polymers such as Eudragit can be used.
- the coating solutions of the present invention preferably contain, in addition to the film-former, plasticizer, and solvent system (i.e., water), a colorant to provide elegance and product distinction.
- Color may be added to the solution of the therapeutically active agent instead, or in addition to the aqueous dispersion of hydrophobic material.
- color may be added to Aquacoat via the use of alcohol or propylene glycol based color dispersions, milled aluminum lakes and opacif ⁇ ers such as titanium dioxide by adding color with shear to water soluble polymer solution and then using low shear to the plasticized Aquacoat.
- any suitable method of providing color to the formulations of the present invention may be used.
- Suitable ingredients for providing color to the formulation when an aqueous dispersion of an acrylic polymer is used include titanium dioxide and color pigments, such as iron oxide pigments. The incorporation of pigments, may, however, increase the retard effect of the coating.
- Plasticized hydrophobic material may be applied onto the substrate comprising the therapeutically active agent by spraying using any suitable spray equipment known in the art.
- a Wurster fluidized- bed system is used in which an air jet, injected from underneath, fluidizes the core material and effects drying while the acrylic polymer coating is sprayed on.
- a further overcoat of a film-former such as Opadry, is optionally applied to the beads. This overcoat is provided, if at all, in order to substantially reduce agglomeration of the beads.
- the release of the therapeutically active agent from the controlled release formulation of the present invention can be further influenced, i.e., adjusted to a desired rate, by the addition of one or more release-modifying agents, or by providing one or more passageways through the coating.
- the ratio of hydrophobic material to water soluble material is determined by, among other factors, the release rate required and the solubility characteristics of the materials selected.
- the release-modifying agents which function as pore-formers may be organic or inorganic, and include materials that can be dissolved, extracted or leached from the coating in the environment of use.
- the pore-formers may comprise one or more hydrophilic materials such as hydroxypropyl methylcellulose.
- the sustained release coatings of the present invention can also include erosion-promoting agents such as starch and gums.
- the sustained release coatings of the present invention can also include materials useful for making microporous lamina in the environment of use, such as polycarbonates comprised of linear polyesters of carbonic acid in which carbonate groups reoccur in the polymer chain.
- the release-modifying agent may also comprise a semi-permeable polymer.
- the release-modifying agent is selected from hydroxypropyl methylcellulose, lactose, metal stearates, and mixtures of any of the foregoing.
- the sustained release coatings of the present invention may also include an exit means comprising at least one passageway, orifice, or the like.
- the passageway may be formed by such methods as those disclosed in U.S. Pat. Nos. 4,063,064 and 4,088,864 (all of which are hereby incorporated by reference).
- the passageway can have any shape such as round, triangular, square, elliptical, irregular, etc.
- the controlled release formulation is achieved via a matrix having a controlled release coating as set forth above.
- the present invention also comprises sustained-release tablets comprising an opioid agonist and cannabinoid antagonist particles coated with a coating that renders the antagonist substantially non-releasable, wherein the agonist and the antagonist are dispersed in a controlled release matrix that affords in-vitro dissolution rates of the opioid agonist within the preferred ranges and that releases the opioid agonist in a pH-dependent or pH- independent manner.
- the materials suitable for inclusion in a controlled release matrix will depend on the method used to form the matrix.
- a matrix in addition to the opioid agonist and the substantially non-releasable form of the coated cannabinoid antagonist may include: (i) Hydrophilic and/or hydrophobic materials, such as gums, cellulose ethers, acrylic resins, protein derived materials; the list is not meant to be exclusive, and any pharmaceutically acceptable hydrophobic material or hydrophilic material which is capable of imparting controlled release of the cannabinoid may be used in accordance with the present invention; (2) Digestible, long chain (C 8 -C50, especially Ci 2 -C 40 ), substituted or unsubstituted hydrocarbons, such as fatty acids, fatty alcohols, glyceryl esters of fatty acids, mineral and vegetable oils and waxes, and stearyl alcohol; and polyalkylene glycols.
- Hydrophilic and/or hydrophobic materials such as gums, cellulose ethers, acrylic resins, protein derived materials; the list is not meant to be exclusive, and any pharmaceutically acceptable hydrophobic material or hydrophil
- the oral dosage form may contain between 1% and 80% (by weight) of at least one hydrophilic or hydrophobic material.
- the hydrophobic material is a hydrocarbon
- the hydrocarbon preferably has a melting point of between 25°C and 90°C.
- fatty (aliphatic) alcohols are preferred.
- the oral dosage form may contain up to 60% (by weight) of at least one digestible, long chain hydrocarbon.
- the oral dosage form contains up to 60% (by weight) of at least one polyalkylene glycol.
- the hydrophobic material is preferably selected from the group consisting of alkylcelluloses, acrylic and methacrylic acid polymers and copolymers, shellac, zein, hydrogenated castor oil, hydrogenated vegetable oil, or mixtures thereof.
- the hydrophobic material is a pharmaceutically acceptable acrylic polymer, including but not limited to acrylic acid and methacrylic acid copolymers, methyl methacrylate, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, aminoalkyl methacrylate copolymer, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamine copolymer, poly(methyl methacrylate), poly(methacrylic acid)(anhydride), polymethacrylate, polyacrylamide, poly(methacrylic acid anhydride), and glycidyl methacrylate copolymers.
- the hydrophobic material is selected from materials such as hydroxyalkylcelluloses such as hydroxypropylmethylcellulose and mixtures of the foregoing.
- hydrophobic materials are water-insoluble with more or less pronounced hydrophilic and/or hydrophobic trends.
- the hydrophobic materials useful in the invention have a melting point from about 3O 0 C to about 200 0 C, preferably from about 45°C to about 90 0 C.
- the hydrophobic material may comprise natural or synthetic waxes, fatty alcohols (such as lauryl, myristyl, stearyl, cetyl or preferably cetostearyl alcohol), fatty acids, including but not limited to fatty acid esters, fatty acid glycerides (mono-, di-, and ⁇ -glycerides), hydrogenated fats, hydrocarbons, normal waxes, stearic aid, stearyl alcohol and hydrophobic and hydrophilic materials having hydrocarbon backbones.
- Suitable waxes include, for example, beeswax, glycowax, castor wax and camauba wax.
- a wax-like substance is defined as any material which is normally solid at room temperature and has a melting point of from about 3O 0 C to about 100 0 C.
- Suitable hydrophobic materials which may be used in accordance with the present invention include digestible, long chain (C 8 -C 5O , especially Ci 2 - C 40 ), substituted or unsubstituted hydrocarbons, such as fatty acids, fatty alcohols, glyceryl esters of fatty acids, mineral and vegetable oils and natural and synthetic waxes. Hydrocarbons having a melting point of between 25 0 C and 9O 0 C are preferred. Of the long chain hydrocarbon materials, fatty (aliphatic) alcohols are preferred in certain embodiments.
- the oral dosage form may contain up to 60% (by weight) of at least one digestible, long chain hydrocarbon.
- a combination of two or more hydrophobic materials are included in the matrix formulations.
- an additional hydrophobic material is included, it is preferably selected from natural and synthetic waxes, fatty acids, fatty alcohols, and mixtures of the same. Examples include beeswax, camauba wax, stearic acid and stearyl alcohol. This list is not meant to be exclusive.
- One particular suitable matrix comprises at least one water soluble hydroxyalkyl cellulose, at least one Ci 2 -C 36 , preferably CH-C 22 , aliphatic alcohol and, optionally, at least one polyalkylene glycol.
- the at least one hydroxyalkyl cellulose is preferably a hydroxy (Ci to C 6 ) alkyl cellulose, such as hydroxypropylcellulose, hydroxypropylmethylcellulose and, especially, hydroxyethylcellulose.
- the amount of the at least one hydroxyalkyl cellulose in the present oral dosage form will be determined, inter alia, by the precise rate of cannabinoid release required.
- the at least one aliphatic alcohol may be, for example, lauryl alcohol, myristyl alcohol or stearyl alcohol. In particularly preferred embodiments of the present oral dosage form, however, the at least one aliphatic alcohol is cetyl alcohol or cetostearyl alcohol.
- the amount of the at least one aliphatic alcohol in the present oral dosage form will be determined, as above, by the precise rate of cannabinoid release required. It will also depend on whether at least one polyalkylene glycol is present in or absent from the oral dosage form. In the absence of at least one polyalkylene glycol, the oral dosage form preferably contains between 20% and 50% (by wt) of the at least one aliphatic alcohol. When at least one polyalkylene glycol is present in the oral dosage form, then the combined weight of the at least one aliphatic alcohol and the at least one polyalkylene glycol preferably constitutes between 20% and 50% (by wt) of the total dosage.
- the ratio of, e.g., the at least one hydroxyalkyl cellulose or acrylic resin to the at least one aliphatic alcohol/polyalkylene glycol determines, to a considerable extent, the release rate of the cannabinoid from the formulation.
- a ratio of the at least one hydroxyalkyl cellulose to the at least one aliphatic alcohol/polyalkylene glycol of between 1 :2 and 1 :4 is preferred, with a ratio of between 1 :3 and 1 :4 being particularly preferred.
- the at least one polyalkylene glycol may be, for example, polypropylene glycol or, which is preferred, polyethylene glycol.
- the number average molecular weight of the at least one polyalkylene glycol is preferred between 1,000 and 15,000 especially between 1,500 and 12,000.
- Another suitable controlled release matrix would comprise an alkylcellulose (especially ethyl cellulose), a Ci 2 to C 36 aliphatic alcohol and, optionally, a polyalkylene glycol.
- the matrix includes a pharmaceutically acceptable combination of at least two hydrophobic materials.
- a controlled release matrix may also contain suitable quantities of other materials, e.g. diluents, lubricants, binders, granulating aids, colorants, flavorants and glidants that are conventional in the pharmaceutical art.
- any method of preparing a matrix formulation known to those skilled in the art may be used.
- incorporation in the matrix may be effected, for example, by (a) forming granules comprising at least one water soluble hydroxyalkyl cellulose and cannabinoid or a cannabinoid salt; (b) mixing the hydroxyalkyl cellulose containing granules with at least one Ci 2 -C 36 aliphatic alcohol; and (c) optionally, compressing and shaping the granules.
- the granules are formed by wet granulating the hydroxy-alkyl cellulose/cannabinoid with water.
- the amount of water added during the wet granulation step is preferably between 1.5 and 5 times, especially between 1.7"5 and 3.5 times, the dry weight of the cannabinoid.
- a spheronizing agent together with the active ingredient can be spheronized to form spheroids.
- Macrocrystalline cellulose is preferred.
- a suitable microcrystalline cellulose is, for example, the material sold as Avicel PH 101.
- the spheroids may also contain a binder. Suitable binders, such as low viscosity, water soluble polymers, will be well known to those skilled in the pharmaceutical art. However, water soluble hydroxy lower alkyl cellulose, such as hydroxypropylcellulose, are preferred.
- the spheroids may contain a water insoluble polymer, especially an acrylic polymer, an acrylic copolymer, such as a methacrylic acid-ethyl acrylate copolymer, or ethyl cellulose.
- the sustained release coating will generally include a hydrophobic material such as (a) a wax, either alone or in admixture with a fatty alcohol; or (b) shellac or zein.
- Sustained release matrices can also be prepared via melt-granulation or melt-extrusion techniques, as long as the techniques used do not damage the integrity of the substantially non-releasable form of the cannabinoid antagonist added during the preparation of the matrix to the extent that sufficient amount of the cannabinoid antagonist becomes available to be released into the gastrointestinal system upon oral administration.
- the melt extrusion step may be performed with the opioid agonist to produce sustained release particles of the agonist, which may then be combined with the substantially non-releasable form of the cannabinoid antagonist.
- melt-granulation techniques involve melting a normally solid hydrophobic material, e.g. a wax, and incorporating a powdered drug therein.
- sustained release dosage form it may be necessary to incorporate an additional hydrophobic substance, e.g. ethylcellulose or a water-insoluble acrylic polymer, into the molten wax hydrophobic material.
- additional hydrophobic substance e.g. ethylcellulose or a water-insoluble acrylic polymer
- sustained release formulations prepared via melt-granulation techniques are found in U.S. Pat. No. 4,861,598, and hereby incorporated by reference in its entirety.
- the additional hydrophobic material may comprise one or more water- insoluble wax-like thermoplastic substances possibly mixed with one or more wax-like thermoplastic substances being less hydrophobic than said one or more water-insoluble wax-like substances.
- the individual wax-like substances in the formulation should be substantially non-degradable and insoluble in gastrointestinal fluids during the initial release phases.
- Useful water-insoluble wax-like substances may be those with a water-solubility that is lower than about 1 : 5,000 (w/w).
- a sustained release matrix may also contain suitable quantities of other materials, e.g., diluents, lubricants, binders, granulating aids, colorants, flavorants and glidants that are conventional in the pharmaceutical art. The quantities of these additional materials will be sufficient to provide the desired effect to the desired formulation.
- a sustained release matrix incorporating melt-extruded multiparticulates may also contain suitable quantities of other materials, e.g. diluents, lubricants, binders, granulating aids, colorants, flavorants and glidants that are conventional in the pharmaceutical art in amounts up to about 50% by weight of the particulate if desired.
- the preparation of a suitable melt-extruded matrix according to the present invention may, for example, include the steps of blending the cannabinoid analgesic, together with at least one hydrophobic material and preferably the additional hydrophobic material to obtain a homogeneous mixture.
- the homogeneous mixture is then heated to a temperature sufficient to at least soften the mixture sufficiently to extrude the same.
- the resulting homogeneous mixture is then extruded to form strands.
- the extrudate is preferably cooled and cut into multiparticulates by any means known in the art.
- the strands are cooled and cut into multiparticulates.
- the multiparticulates are then blended with the cannabinoid antagonist particles coated with a coating that renders the antagonist substantially non-releasable and divided into unit doses.
- the extrudate preferably has a diameter of from about 0.1 to about 5 mm and provides sustained release of the opioid agonist for a time period of from about 8 to about 24 hours.
- An optional process for preparing the melt extrusions of the present invention includes directly metering into an extruder a hydrophobic material, a therapeutically active agent, and an optional binder; heating the homogenous mixture; extruding the homogenous mixture to thereby form strands; cooling the strands containing the homogeneous mixture; cutting the strands into particles having a size from about 0.1 mm to about 12 mm; and combining the particles with the coated cannabinoid antagonist particles and dividing them into unit doses.
- a relatively continuous manufacturing procedure is realized.
- the diameter of the extruder aperture or exit port can also be adjusted to vary the thickness of the extruded strands.
- the exit part of the extruder need not be round; it can be oblong, rectangular, etc.
- the exiting strands can be reduced to particles using a hot wire cutter, guillotine, etc.
- the melt extruded multiparticulate system can be, for example, in the form of granules, spheroids or pellets depending upon the extruder exit orifice.
- the terms "melt-extruded multiparticulate(s)” and “melt-extruded multiparticulate system(s)” and “melt- extruded particles” shall refer to a plurality of units, preferably within a range of similar size and/or shape and containing one or more active agents and one or more excipients, preferably including a hydrophobic material as described herein.
- melt-extruded multiparticulates will be of a range of from about 0.1 to about 12 mm in length and have a diameter of from about 0.1 to about 5 mm.
- melt-extruded multiparticulates can be any geometrical shape within this size range.
- the extrudate may simply be cut into desired lengths and divided into unit doses of the therapeutically active agent without the need of a spheronization step.
- oral dosage forms are prepared to include an effective amount of melt-extruded multiparticulates within a capsule.
- a plurality of the melt-extruded multiparticulates may be placed in a gelatin capsule in an amount sufficient to provide an effective sustained release dose when ingested and contacted by gastric fluid.
- a suitable amount of the multiparticulate extrudate is combined with the coated cannabinoid antagonist particles and compressed into an oral tablet using conventional tableting equipment using standard techniques. Techniques and compositions for making tablets (compressed and molded), capsules (hard and soft gelatin) and pills are also described in Remington's Pharmaceutical Sciences, incorporated by reference herein.
- the coated cannabinoid antagonist particles are added during the extrusion process and the extrudate can be shaped into tablets as set forth in U.S. Pat. No. 4,957,681, described in additional detail above and hereby incorporated by reference.
- the sustained release melt-extruded multiparticulate systems or tablets can be coated, or the gelatin capsule can be further coated, with a sustained release coating such as the sustained release coatings described above.
- a sustained release coating such as the sustained release coatings described above.
- Such coatings preferably include a sufficient amount of hydrophobic material to obtain a weight gain level from about 2 to about 30 percent, although the overcoat may be greater depending upon the physical properties of the particular cannabinoid analgesic compound utilized and the desired release rate, among other things.
- the melt-extruded unit dosage forms of the present invention may further include combinations of melt-extruded multiparticulates containing one or more of the therapeutically active agents disclosed above before being encapsulated. Furthermore, the unit dosage forms can also include an amount of an immediate release opioid agonist for prompt therapeutic effect.
- the immediate release opioid agonist may be incorporated, e.g., as separate pellets within a gelatin capsule, or may be coated on the surface of the multiparticulates after preparation of the dosage forms (e.g., controlled release coating or matrix-based).
- the unit dosage forms of the present invention may also contain a combination of controlled release beads and matrix multiparticulates to achieve a desired effect.
- the sustained release formulations of the present invention preferably slowly release the opioid agonist, e.g., when ingested and exposed to gastric fluids, and then to intestinal fluids.
- the sustained release profile of the melt- extruded formulations of the invention can be altered, for example, by varying the amount of retardant, i.e., hydrophobic material, by varying the amount of plasticizer relative to hydrophobic material, by the inclusion of additional ingredients or excipients, by altering the method of manufacture, etc.
- the melt extruded material is prepared without the inclusion of the opioid agonist and/or coated cannabinoid antagonist particles, which are added thereafter to the extrudate.
- Such formulations typically will have the drugs blended together with the extruded matrix material, and then the mixture would be tableted in order to provide a slow release of the opioid agonist.
- Such formulations may be advantageous, for example, when the therapeutically active agent included in the formulation is sensitive to temperatures needed for softening the hydrophobic material and/or the retardant material.
- the dosage form when the non-releasable aversive agent is a liquid, or a semisolid or a poorly water soluble liquid or solid, the dosage form may be prepared using other methods.
- the dosage form may be mixed and blended, optionally in the presence of heat, with a fine powder excipient (e.g., talc), the blended material continuously fed into a twin screw extruder, and the resultant strands collected on a conveyor.
- a fine powder excipient e.g., talc
- the strands are allowed to cool on the conveyor and then cut into pellets using a Pelletizer.
- the the pellets are screened and' the desired sieve portion collected.
- the water is replaced with ethanol, hydroalcoholic solutions or organic solvents know in the art to aid in the dissolution, dispersion or mixing of the aversive agent.
- solid dispersions of the aversive agent and HPMC are prepared by hot-stage extrusion with a corotating twin-screw extruder.
- the extrudates are collected after cooling at ambient temperature on a conveyor belt. Samples are milled for 1 min with a laboratory cutting mill and sieved to the desired particle size.
- sequestered agent selected from the group comprising cannabinoid antagonists, alcohol deterrents, and mixture thereof, as defined in this invention may also be prepared by modification of the examples herein and by use of material other than those specifically disclosed herein, including those which may hereafter become known to the art to be capable of performing the necessary functions.
- More than one substantially non-releasable aversive agent selected from the group comprising cannabinoid antagonists and alcohol deterrents may be included in the dosage form either in the form of separate substantially non-releasable subunits, on combined into the same substantially non- releasable subunits.
- Said more than one substantially non-releasable aversive agent may comprise aversive agents from the same class of agents (e.g., more than one cannabinoid antagonist, or more than one alcohol deterrent) or from a different class of agents (e.g., one cannabinoid antagonist, one alcohol deterrent) or mixtures thereof.
- the percent loading of the opioid agonist agent, the substantially non- releasable aversive agent and the abuse intervention agent onto the beads may be varied depending on the physiochemical and pharmaceutical properties of said agent and ingredients (excipients), the pharmacologic effects of said agent and the desired degree of release or non-release from the dosage form.
- the aversive agent When the aversive agent is in the form of multiparticulates, including beads and granulation individually coated with a sequestering material, it may be directly or indirectly be overcoated with the opioid agonist such that each multiparticulate contains a aversive agent and an opioid agonist and filled uncompressed into a capsule or compressed into a capsule or tablet.
- the ingredients used for the preparation of the releasable opioid agonist in immediate release form or controlled release form, and the substantially non-releasable aversive agent may be modified depending on the selection, dose and desired duration of effect of the opioid agonist and the aversive agent.
- a change in the dose or amount opioid agonist and/or aversive agent does not require a change in amount of other ingredients.
- a proportional change in the amount of other ingredients is required to maintain the desired properties.
- a change in the dose or amount opioid agonist and/or aversive agent necessitates a change in the nature and/or amount of ingredients to provide the required characteristics of the opioid agonist (e.g., immediate release, sustained release, duration of effect, rate and extent of absorption, therapeutic concentrations and effect, etc.) and aversive agent (e.g., extent of non-release or sequestration, degree of abuse deterrence, etc).
- opioid agonist e.g., immediate release, sustained release, duration of effect, rate and extent of absorption, therapeutic concentrations and effect, etc.
- aversive agent e.g., extent of non-release or sequestration, degree of abuse deterrence, etc.
- Abuse intervention agents may optionally be incorporated into the same sub-unit as the substantially non-releasable aversive agent or into a different sub-unit or into the granulation or matrix material containing the opioid agonist.
- the ingredients used for the preparation of the releasable opioid agonist in immediate release form or controlled release form, and the substantially non-releasable aversive agent may be used or further modified for the preparation of in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form abuse intervention agents selected from the group comprising (i) laxatives; (ii) cutaneous vasodilators; (iii) headache producing agents; (iv) emetics, emetogenic and nausea producing compounds; (iv) bittering agents (v) mucosal, naso-mucosal, oro-mucosal, respiratory, tissue and gastrointestinal irritants; (vi) tissue staining, non-tissue staining and beverage staining dyes, lakes and colorants; (vii) fecal and urine discolorants; (viii) malodorous agents; (ix) opioid antagonists;
- Example 1 to 76 a substantially non-releasable forms of aversive agents (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents and mixtures thereof is prepared by coating or mixing said aversive agent with a material that renders it substantially non-releasable.
- aversive agents i.e., sequestered agent
- Aversive Agent Loading on Sphere Add Rimonabant HCl in ethanol with stirring, then add Opadry White and continue mixing until a homogenous dispersion is produced. Apply the above dispersion onto the sugar spheres using a fluid bed coater.
- Sphere Overcoating Disperse Opadry White in purified water with stirring and apply the dispersion over the sugar spheres loaded with Rimonabant HCl using a fluid bed coater.
- Sequestration Overcoating Mix Eudragit RS30d, triethyl citrate, talc and purified water with stirring and apply the dispersion over the loaded and overcoated sugar spheres using a fluid bed coater (i.e., a fluid bed coating machine).
- Final Coating Disperse Opadry White in purified water with stirring and apply the dispersion foregoing (sequestration overcoated product).
- Cure Cure the foregoing spheres at 45 ° for approximately 48 to 72 hours.
- Example 2 is prepared as in Example 1, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-rnethyl-lH-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM
- Example 3 is prepared as in Example 1, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Aversive Agent Pellets The Gelucire 44/14 is dispensed into a mixer and heated until fully melted. The hydroxypropyl methyl cellulose (HPMC) is dispensed into the mixer. The mixing is continued until fully dispersed. The Rimonabant is dispensed into the same vessel. The mixture is thoroughly dispersed with a high shear mixer. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The Conveyor is set to provide extrudate diameter of 0.5 mm or 1 mm and the Pelletizer is set to provide pellets of approximately 0.5 or 1 mm length.
- Pellet Overcoating The Opadry White is dispersed in purified water with stirring and the dispersion applied over the pellets containing Rimonabant HCl using a fluid bed coater.
- Sequestration Overcoating The Eudragit RS30d, triethyl citrate, talc and purified water are mixed with stirring and applied over the loaded and overcoated pellets using a fluid bed coater (i.e., a fluid bed coating machine).
- Final Coating The Opadry White is dispersed in purified water with stirring and the dispersion is applied to the foregoing (sequestration overcoated product).
- Cure The foregoing pellets are cured at 45 ° for approximately 48 to 72 hours.
- Example 4 is prepared as in Example 1, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-rnethylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630
- Example 5 is prepared as in Example 1, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 6 is prepared as in Example 1, except that the HPMC is replaced with an equal amount of further Gelucire 44/14.
- Example 7 is prepared as in Example 6, except that the Rimonabant
- HCI 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308,
- Example 8 is prepared as in Example 6, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 9 is prepared as in Example 1, except that the Gelucire 44/14 is replaced with an equal amount of Labrafil M 2130 CS.
- Example 10 is prepared as in Example 9, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising * AM251, AM281 ([N-m ⁇ holin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)- 1 -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630
- Example 1 1 are prepared as in Example 9, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymo ⁇ hs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymo ⁇ hs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric
- Example 12 is prepared as in Example 9, except that the HPMC is replaced with an equal amount of further Labrafil M 2130 CS.
- Example 13 is prepared as in Example 12, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU
- Example 14 is prepared as in Example 12, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 15 is prepared as in Example 1, except that the Gelucire
- Example 16 is prepared as in Example 15, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308,
- Example 17 is prepared as in Example 15, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymo ⁇ hs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymo ⁇ hs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric
- Example 18 is prepared as in Example 15, except that the HPMC is replaced with an equal amount of further Beeswax.
- EXAMPLE 19 is prepared as in Example 15, except that the HPMC is replaced with an equal amount of further Beeswax.
- Example 19 is prepared as in Example 18, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308,
- Example 20 is prepared as in Example 18, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymo ⁇ hs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymo ⁇ hs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric
- Example 21 is prepared as in Example 1, except that the Gelucire
- Example 22 is prepared as in Example 21, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SRl 44528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630
- Example 23 is prepared as in Example 21, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 24 is prepared as in Example 1, except that the HPMC is replaced with an equal amount of further Cithrol GMS.
- Example 25 is prepared as in Example 24, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxarnide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SRl 41716, SR 144528, AM630,
- Example 26 is prepared as in Example 24, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 27 is prepared as in Example 1, except that the Gelucire
- Example 28 is prepared as in Example 27, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SRl 44528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR 144528, AM630, HU
- Example 30 is prepared as in Example 27, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 30 is prepared as in Example 27, except that the HPMC is replaced with an equal amount of further Hydrokote 1 12.
- Example 31 is prepared as in Example 27, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630,
- Example 32 is prepared as in Example 27, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 33 is prepared as in Example 1, except that the Gelucire
- Example 34 is prepared as in Example 33, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-rnethyl-lH-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methy 1-pheny I)- 1 -(4-methy lbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716,
- Example 35 is prepared as in Example 33, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 36 is prepared as in Example 33, except that the HPMC is replaced with an equal amount of further glyceryl behenate (CompitrolTM 888 ATO).
- Example 37 is prepared as in Example 33, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-mo ⁇ holin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, SR141716, SR144528, AM630, HU
- Example 38 is prepared as in Example 33, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 39 is prepared as in Example 1, except that the Gelucire
- Example 40 is prepared as in Example 39, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[( 1 S)-endo- 1 ,3,3-trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR 144528, AM630,
- Example 41 is prepared as in Example 39, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 42 is prepared as in Example 39, except that the HPMC is replaced with an equal amount of further glyceryl palmitostearate (PrecirolTM ATO 5).
- Example 43 is prepared as in Example 39, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SRl 44528 ([N- ⁇ l S ⁇ endo-l ⁇ -trimethylbicyc ⁇ . ⁇ heptan ⁇ -y ⁇ -chloro-S- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4
- Example 44 is prepared as in Example 39, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 45 is prepared as in any of Examples 6, 12, 18, 24, 30, 36 and
- Example 46 is prepared as in Example 45, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methy 1-pheny I)- 1 -(4-methy lbenzy l)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexy 1- 1 h- 1 ,2,4-triazole, SRl 41716, SR1445
- Example 47 is prepared as in Example 45, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Powder Preparation Dissolve the Rimonabant HCl in chloroform or a suitable alternative organic solvent with mixing. Dissolve PVP in the Rimonabant HCl/chloroform mixture with mixing. Evaporate the chloroform under vacuum, optionally with heating or using a suitable alternative method (e.g., by freeze-drying or by spray-drying) to produce a residual Rimonabant/PVP powder. Micronize the Rimonabant/PVP powder.
- Aversive Agent Loading on Sphere Add Rimonabant HC1/PVP in ethanol with stirring, then add Opadry White and continue mixing until a homogenous dispersion is produced.
- Sphere Overcoating Disperse Opadry White in purified water with stirring and apply the dispersion over the sugar spheres loaded with Rimonabant HCl using a fluid bed coater.
- Sequestration Overcoating Mix Eudragit RS30d, triethyl citrate, talc and purified water with stirring and apply the dispersion over the loaded and overcoated sugar spheres using a fluid bed coater (i.e., a fluid bed coating machine).
- Final Coating Disperse Opadry White in purified water with stirring and apply the dispersion foregoing (sequestration overcoated product).
- Cure Cure the foregoing spheres at 45 ° for approximately 48 to 72 hours.
- Example 49 is prepared as in Example 48, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxarnide), AM630, SR144528 ([N-[( 1 S)-endo- 1 ,3,3-trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630,
- Example 50 is prepared as in Example 49, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Aversive Agent Loading on Sphere Add Rimonabant HCl in purified water with stirring, then add Opadry White and continue mixing until a homogenous dispersion is produced. Apply the above dispersion onto the sugar spheres using a fluid bed coater.
- Sphere Overcoating Disperse Opadry White in purified water with stirring and apply the dispersion over the sugar spheres loaded with rimonabant HCl using a fluid bed coater.
- Sequestration Overcoating Mix Eudragit RS30d, triethyl citrate, talc and purified water with stirring and apply the dispersion over the loaded and overcoated sugar spheres using a fluid bed coater (i.e., a fluid bed coating machine).
- Final Coating Disperse Opadry White in purified water with stirring and apply the dispersion foregoing (sequestration overcoated product).
- Cure Cure the foregoing spheres at 45 ° for approximately 48 to 72 hours.
- Example 52 is prepared as in Example 51, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-l H-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-30
- Example 53 is prepared as in Example 51, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Eudragit RSlOO in ethyl alcohol and dissolving the triethyl citrate in the solution. The Rimonabant HCl cores (700 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed, in a fluid- bed equipped with a Wurster insert.
- Example 55 is prepared as in Example 54, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl- 1 H-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU
- Example 56 is prepared as in Example 54, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Eudragit NE 30D in ethyl alcohol and dissolving the triethyl citrate in the solution. The Rimonabant HCl cores (70 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed in a fluid- bed equipped with a Wurster insert.
- Example 58 is prepared as in Example 57, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-l H-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308,
- Example 59 is prepared as in Example 57, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 60 is prepared as in any of Example 57, Example 58, or
- Example 59 except that the Purified water is replaced with 70% ethanol in water.
- Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Ethylcellulose Nl O in ethyl alcohol and dissolving the dibutyl sebacate in the solution. The Rimonabant HCl cores (70 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed in a fluid-bed equipped with a Wurster insert. EXAMPLE 62
- Example 62 is prepared as in Example 61, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630,
- Example 63 is prepared as in Example 61, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixture
- Example 64 is prepared as in any of Example 61, Example 62, or
- Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Eudragit RSlOO in ethyl alcohol and dissolving the dibutyl sebacate in the solution. The Rimonabant HCl cores (1 15 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed in a fluid- bed equipped with a Wurster insert.
- Example 66 is prepared as in Example 65, except that the Rimonabant
- HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308,
- Example 67 is prepared as in Example 65, except that the Rimonabant
- HCI 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures
- Example 68 is prepared as in any of Example 65, Example 66, or
- Example 67 except that the Purified water is replaced with 70% ethanol in water.
- Rimonabant HCl is dispersed in a hydroalcoholic solution of hypromellose with stirring. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of ethylcellulose and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert. EXAMPLE 70
- Rimonabant HCl is dispersed in a hydroalcoholic solution of hypromellose with stirring. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of Eudragit RS, sodium lauryl sulfate and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
- Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose using a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of ethylcellulose and dibutyl sebacate in ethanol is prepared, which is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
- Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose using a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of ethylcellulose in ethanol is prepared and magnesium stearate is dispersed just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
- Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose with a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of Eudragit RS, sodium lauryl sulfate and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl in Glatt GPCG-3 with a 4" Wurster insert. EXAMPLE 74
- Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose with a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of Eudragit RS, sodium lauryl sulfate and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl in Glatt GPCG-3 with a 4" Wurster insert.
- Example 75 is prepared as in any of Examples 69, 70, 71, 72, 73 or 74, except that the Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4- yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2- yl]5-(4-chloro-3-methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3- carboxamide]), 5-(4-chlorophenyl)-l -(2,4-dichlorophenyl)
- Example 76 is prepared as in any of Examples 69, 70, 71, 72, 73 or 74, except that the Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- aversive agent 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs,
- Example 77 to 99 a substantially non-releasable forms of aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, and alcohol deterrents, or mixtures thereof are prepared by coating aversive agent particles or aversive agent loaded beads with a coating that render it substantially non-releasable.
- aversive agent i.e., sequestered agent
- HCl, Eudragit RSPO and milled stearyl alcohol are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- Eudragit RSPO milled stearyl alcohol, stearic acid and butylated hydroxytoluene are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- Eudragit RSPO milled stearyl alcohol, stearic acid and butylated hydroxytoluene are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- Eudragit RSPO milled stearyl alcohol, stearic acid and butylated hydroxytoluene are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- Eudragit RSPO milled stearyl alcohol, dibasic calcium phosphate and butylated hydroxytoluene are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- Eudragit RSPO milled stearyl alcohol, dibasic calcium phosphate and butylated hydroxytoluene are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- Eudragit RSPO milled stearyl alcohol, dibasic calcium phosphate and butylated hydroxytoluene are mixed in a twin shell blender.
- the blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool.
- the cooled strands are cut into pellets using a pelletizer.
- the pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
- the Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours. The resulting bead formulation can be incorporated into a controlled release Opioid Agonist granulation and the mixture compressed into tablets.
- the Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours.
- the resulting bead formulation can be incorporated into a controlled release Any of example 1 to 99 granulation and the mixture compressed into tablets.
- the Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours.
- the resulting bead formulation can be incorporated into a controlled release Any of example 1 to 99 granulation and the mixture compressed into tablets.
- the stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender.
- the mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer.
- the Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar
- the Conveyor is set to provide extrudate diameter of 1 mm
- the Pelletizer is set to provide pellets of approximately 1 mm length.
- Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
- the stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender.
- the mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer.
- the Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar
- the Conveyor is set to provide extrudate diameter of 1 mm
- the Pelletizer is set to provide pellets of approximately 1 mm length.
- Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
- the stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender.
- the mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer.
- the Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar
- the Conveyor is set to provide extrudate diameter of 1 mm
- the Pelletizer is set to provide pellets of approximately 1 mm length.
- Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
- the stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender.
- the mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer.
- the Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar
- the Conveyor is set to provide extrudate diameter of 1 mm
- the Pelletizer is set to provide pellets of approximately 1 mm length.
- Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
- Examples 94 is prepared in accordance to any of Examples 77 to 93, except that the Rimonabant is first prepared as a powder using the method of Example 48.
- Examples 95 is prepared in accordance to any of Examples 77 to 89, except that in the first step, the dissolution is in a hydroalcoholic solution.
- Examples 96 is prepared in accordance to any of Examples 77 to 89, except that in the first step, the dissolution is in denatured ethyl alcohol.
- Examples 97 is prepared in accordance to any of Examples 77 to 89, except that in the first step, the dissolution is in a suitable organic solvent.
- Example 98 is prepared as in any of Examples 77 to 83, except that the
- Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5- [2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630,
- SR144528 ([N-[(lS)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4- chloro-3-methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric
- Example 99 is prepared as in any of Examples 77 to 83, except that the
- Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
- Example 100 to 1 16 tablet formulation comprising an immediate release opioid agonist with a suitable amount of substantially non-releasable aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents and mixtures thereof is prepared.
- substantially non-releasable aversive agent i.e., sequestered agent
- the preparation of substantially non-releasable aversive agent forms has been described in the forgoing examples.
- the amount of substantially non- releasable aversive agent added to the dosage forms of Example 100 to 1 16 will vary based on a variety of factors previously described.
Abstract
The present invention is directed to pharmaceutical compositions of opioid agonists and the use thereof for preventing or minimizing the risk of abuse and/or toxicity due to opioid agonists and any co-abused cannabinoid agonists or alcohol from either intentional or unintentional tampering. The present invention is also directed at methods of preventing or minimizing the risk of abuse and/or toxicity due to opioid agonists and any co-abused cannabinoid agonists or alcohol from either intentional or unintentional tampering.
Description
ABUSE DETERRENT ORAL PHARMACEUTICAL FORMULATIONS OF OPIOID AGONISTS AND METHOD OF USE
FIELD OF THE INVENTION
[0001] The present invention is in the field of abuse deterrent oral pharmaceutical compositions of opioid agonists and the use thereof.
BACKGROUND OF THE INVENTION
[0002] Currently, medical practitioners may choose from several well- accepted classes of pharmaceutical agents in their attempts to alleviate and prevent pain. Nonlimiting examples of agents used include nonsteroidal antiinflammatory agents (NSAIDs), e.g., aspirin, ibuprofen, ketoprofen, diclofenac; opioids, e.g., morphine, hydromorphone, hydrocodone, levorphanol, oxycodone, tramadol, and codeine; cyclooxygenase-2 (COX-2) selective NSAIDs, e.g., celecoxib, valdecoxib, etoricoxib, lumiracoxib, and rofecoxib; acetaminophen; tricyclic antidepressants, e.g., amitriptyline, despiramine, nortriptyline; non-tricyclic antidepressants, e.g., doxepin, duloxetine, paroxetine, venlafaxine; antiepileptics, e.g., gabapentin, pregabalin, carbamazepine, oxcarbazepine, lamotrigine; voltage sensitive N- type calcium channel blockers, e.g., ziconotide and alpha adrenergic agonists, e.g., clonidine.
[0003] Among the most widely used and effective class of drugs available for pain relief are the opioid agonists such as morphine, hydromorphone, hydrocodone and oxycodone. Opioid agonist are powerful analgesics that provided significant relief from virtually all apjnful conditions know to human. Unfortunately, opioid analgesics are psychoactive substances know to produce iatrogenic addiction in a small nuber of patients with pain. They are also widely sough after by recreational drug users and drug addicts for nonmedical purposes to provide mood altering effects which are perceived to be desireable amongst such recreational drug users and drug addicts.
[0004] Although the abuse of unregulated psychoactive or mood altering naturally occurring and synthetic chemical substances and "street drags" has been documented over many centuries, it is only in the last century that we have been confronted by the abuse of pharmaceutical grade psychoactive or mood altering substances that have been fabricated in the form of pharmaceutical products for therapeutic uses. The abuse of pharmaceutical products is either due to iatrogenic addiction or willful abuse of the products outside their intended use or method of use by drug abusers and recreational drag users. Most of the abuse of pharmaceutical products, regardless of etiology, is confined to substances that have pleasurable, mood altering effect, rewarding and/or reinforcing effect (i.e., abusable drags).
[0005] A wide variety of pharmaceutical products can be subjected to abuse including opioid agonists.
(0006] On its website, the National Institutes of Drag Abuse (NIDA) notes that prescription drags that are abused or used for nonmedical reasons can alter brain activity and lead to dependence. NIDA further cites that commonly abused classes of prescription drags include opioids (often prescribed to treat pain), central nervous system depressants (often prescribed to treat anxiety and sleep disorders), and stimulants (prescribed to treat narcolepsy, ADHD, and obesity). It notes that commonly used opioids include oxycodone (OxyContin™), propoxyphene (Darvon™), hydrocodone (Vicodin), hydromorphone (Dilaudid™), meperidine (Demerol™), and diphenoxylate (Lomotil™). It cautions that long-term use of opioids or central nervous system depressants can lead to physical dependence and addiction (edited for brevity, www.nida.nih.gov, accessed September 7, 2006)
[0007] Opioid agonists are currently in widespread medical use and have been associated with a dramatic increase in use by drag addicts and recreational drag users. Both immediate release and extended release opioids can be abused.
[0008] According to a report by the Associated Press based on analysis of federal drag prescription data, retail sales of the five leading analgesic in the United States increased 90% from 1997 to 2005. Oxycodone, the
pharmaceutical agent in the sustained release product OxyContin™, was responsible for most of the increase. Oxycodone sales increased nearly sixfold between 1997 and 2005.
[0009J An important goal of analgesic therapy is to achieve continuous relief of pain. Regular administration of an analgesic is generally required to ensure that the next dose is given before the effects of the previous dose have worn off. Continuous suppression of pain through the use of around the clock opioid analgesics is now recommended in treatment guidelines (Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain, Fifth Ed., American, Pain Society (2003); Evidence Based Report of the U.S. Agency for Healthcare Research and Quality (AHRQ) on the Management of Cancer Pain, Report No. 35, AHRQ Publication No. 02-E002, October 2001; Carr et al. J Nat Cancer Inst Monograph 2004;32:23-31; Agency for Health Care Policy and Research Clinical Practice Guidelines for Cancer Pain Management, Guideline No. 9, AHCPR Publication No. 94-0592, March 1994; Agency for Health Care Policy and Research Clinical Practice Guideline for Acute Pain Management, Guideline No. 1, AHCPR Publication No. 92- 0032, February, 1992;Guideline for the Management of Cancer Pain in Adults, American Pain Society, 2005; Guideline for the Management of Pain in Osteoarthritis, Rheumatoid Arthritis, and Juvenile Chronic Arthritis, 2nd Ed., American Pain Society, 2002).
[0010] Conventional (so called "immediate-release", "rapid release" or "short acting") opioid analgesics have been demonstrated to provide short-lived plasma levels, thereby requiring dosing every 4-6 hours in chronic pain. In contrast, extended release oral opioids are designed to maintain effective plasma levels throughout a 12 or 24-hour dosing interval. Extended release opioid formulations have now become the standard of care for the management of chronic pain. Use of extended release opioids can result in fewer interruptions in sleep, reduced dependence on caregivers, improved compliance, enhanced quality of life outcomes, and increased control over the management of their pain. In addition, such formulations can provide more constant plasma concentrations and clinical effects, less frequent peak to
trough fluctuations and fewer side effects, compared with short acting opioids (Sloan and Babul, Expert Opinion on Drug Delivery 2006; Babul et al. Journal of Pain and Symptom Management 2004;28:59-71; Matsumoto et al., Pain Medicine 2005;6:357-66; Dhaliwal et al., Journal of Pain Symptom Management 1995;10:612-23; Hays et al., Cancer 1994;74: 1808-16; Arkinstall et al., Painl995;64: 169-78; Hagen et al., Journal of Clinical Pharmacology 1995;35:38-45; Peloso et al., Journal of Rheumatology 2000;27:764-71).
[0011) Several studies have suggested the benefits of extended release over immediate release opioids. Ferrell et al (Oncol Nur Forum 1989; 4:521-6) compared 12-hourly controlled release morphine and short-acting analgesics in cancer pain and demonstrated that compliance increased as the required dosing frequency decreased, and noncompliance resulted in suboptimal pain control and poor quality-of-life outcomes. Arkinstall et al. (Pain 1995;64: 169-78) demonstrated that twice daily administration of controlled release codeine provided superior to pain control than a PRN regimen of acetaminophen plus codeine.
[0012] An important drawback with the use of opioids is the risk of drug addiction, drug diversion and drug abuse. Although the use of opioids for non-medical purposes has existed throughout recorded human history, their abuse has increased significantly in the past two decades (Drug Abuse Warning Network, http://dawninfo.samhsa.gov/; DEA, http://www.deadiversion.usdoj.gov/; National Survey on Drug Use & Health, http://www.oas.samhsa.gov/nhsda.htm; American Association of Poison Control Centers Toxic Exposure Surveillance System, http://www.aapcc.org/annual.htm).
[0013] Our increased understanding of the clinical pharmacology of opioids and data from well controlled clinical trials in chronic non-cancer pain (Peloso et al., Journal of Rheumatology 2000;27: 764-71; Caldwell, et al., Journal of Pain and Symptom Management 2002;23:278-91; Matsumoto et al., Pain Medicine 2005;6:357-66; Arkinstall et al., Painl995;64: 169-78) and neuropathic pain (Watson and Babul, Neurology 1998;50:1837-41) have
resulted in more widespread use in patients with non-malignant pain (for review, see Sloan and Babul, Expert Opinion on Drug Delivery 2006). This in turn has led to concerns about the increased non-medical use of opioids through both licit and illicit channels. For instance, unsuspecting clinicians may prescribe opioids for pain to individuals with an addiction disorder or individuals with pain who divert a portion of their prescribed dose to other individuals. There have also been documented cases of inappropriate prescribing or dispensing of opioids by physicians and pharmacists, with its eventual diversion into the non-medical marketplace. Additionally, nonmedical supplies of pharmaceutical grade opioids are often obtained through prescription forgeries and break-ins into pharmacies.
[0014] Pharmaceutical dosage forms containing opioids have been used for non-medical purposes in a variety of settings: i) by patients with pain who have developed an addiction disorder following initiation of opioid therapy; ii) by patients with pain who had a pre-existing addiction disorder; iii) by patients with an addiction disorder seeking opioids for their mood altreing properties.
[0015] Non-medical users of opioid analgesics are generally either recreational drug users who may use such agents episodically, or individuals with a addiction disorder who may require frequent maintenance doses. Opioid analgesics may be ingested whole, crushed and ingested, crushed or vaporized and snorted or injected intravenously after attempted extraction of the active pharmaceutical ingredient. The manipulation of pharmaceutical dosage forms of opioids has been documented for many decades. For instance, pentazocine (Talwin™), a synthetic opioid was crushed, extracted and injected intravenously by drug addicts. In other settings, opioid agonists in sustained release form may be crushed or otherwise tampered to liberate make the contents immediate release and then ingested. In yet other settings, ipioid agonists may be tampered and then administered by the non-oral route (e.g., parenteral, intranasal and inhalations use). In yet other settings, tampered opioids may be administered with or in the presence of other mood altering licit and illicit mood altering substances and pharmaceutical agents.
[0016] The introduction of extended release morphine (MS Contin™) revolutionized the management of cancer pain. MS Contin™ gained widespread acceptance due to its global availability, significant pharmacokinetic and pharmacodynamic data, and the convenience of a extended-release formulation. However, the incidence and severity of side effects limits the use of morphine in some patients (Hagen and Babul, Cancer 1997;79: 1428-37). In patients with renal impairment, morphine's principal metabolites, morphine-3- glucuronide and morphine-6-glucuronide can accumulate. Morphine-3-- glucuronide accumulation has been implicated in hyperalgesia, respiratory stimulation, and behavioral excitatory properties through nonopioid receptor mechanisms. Morphine-6-glucuronide accumulation has been implicated in increasing levels of nausea and sedation in patients with renal impairment (Babul and Darke, Clin Pharm Ther, 1993;54:286-92).
[0017] Clinicians treating cancer pain with opioids have reported significant variability among patients in efficacy and side effects with available opioid analgesics. Patients with poor analgesic efficacy or safety outcomes on one opioid frequently tolerate another opioid well. This clinical observation led to the development of oxycodone ER (OxyContin™). Due to the limitations associated with extended release morphine noted above and the "stigma" associated with its use (i.e., association with addiction, advanced cancer, dying and death), extended release oxycodone gained rapid acceptance by patients with chronic non-cancer pain. However, its widespread use for the treatment of chronic non-malignant pain was also associated with its diversion into the non-medical supply for use both by addicts and recreational drug users.
[0018] The popularity of extended release oxycodone among addicts and recreational drug users is at least in part due to a large amount of drug per tablet (a 12 or 24 hour supply). Commercially available immediate release opioid tablets and capsules are usually administered every 4 to 6 hours and they release their dose into the systemic circulation over one to two hours. New, extended release formulations are designed to gradually release their much larger opioid content over a 12 or 24-hour period. Most recreational
drug users and addicts have a unit of use which is one tablet or capsule. The 12 or 24-hour supply of opioid contained in one tablet or capsule, instead of 4 to 6 tablets or capsules means that there is a greater risk that such formulations may be highly sought by drug addicts and recreational drug users alike, for non-medical use. Intentional or inadvertent tampering from extended release formulations will rapidly deliver a massive dose and produce profound a variety of serious and life threatening side effects, including respiratory depression and failure, sedation, cardiovascular collapse, coma and death.
[0019] Addicts and recreational drug users commonly use extended release opioids by a variety of routes of administration. Commonly used methods include 1) parenteral (e.g., intravenous injection), 2) intranasal (e.g., snorting), 3) episodic or repeated oral ingestion of intact, crushed or otherwise tampered tablets or capsules, and 4) in the setting of poly drug abuse (e.g., with co-used or co-abused licit or illicit mood altering or abusable agents).
[0020] One mode of abuse involves the extraction of the opioid component from the dosage form by first mixing the table or capsule with a suitable solvent (e.g., water or alcohol), and then filtering and/or extracting the opioid component from the mixture fqr intravenous injection. Another mode of abuse of extended release opioids involves dissolving the drug in water, alcohol or another "recreational solvent" to hasten its release and to ingest the contents orally, in order to provide high peak concentrations and maximum euphoriant effects.
[0021] A number of strategies have been introduced to minimize the abuse of mood altering drugs. Primary among these schemes is a legal infrastructure that controls the manufacture, distribution and sale of such drugs. In the United States, the vast majority of opioid drugs having clinically useful and approved effects are restricted to dispensing on a prescription-only basis. Most of these drugs are "scheduled" as "controlled drugs", such that distribution of the drug is subject to strict controls and overview. The idea behind scheduling opioid drugs as "controlled" is to ensure that the drugs are dispensed only for the amelioration of legitimate therapeutic maladies, and not for any mood-altering
effect "high" or euphoria that may be produced by the drug when used in supra- therapeutic doses or administered by non-approved routes of administration.
[0022] While the scheduling of opioids as "controlled drugs" has reduced abuse of the drugs, it has not been entirely successful. For example, some persons who are legitimately prescribed the drugs sometimes divert the drugs to persons seeking their procurement for "recreational uses." These "recreational drug users" are frequently willing to pay significant sums of money for the drugs. In other cases, certain health professionals, unfortunately, have been found to be culprits in the non-approved distribution of opioid drugs.
[0023] It is believed that the most widely used diversion techniques at the
"street level" are "doctor shopping" and prescription forgeries. In the case of the former, individuals who may or may not have a legitimate ailment requiring a doctor's prescription for controlled substances, visit numerous doctors, sometimes in several states, to acquire large amounts of controlled substances they abuse or sell to others.
[0024] Scheduling of opioid drugs has also had the unintentional side-effect of causing physicians, fearful of being accused of permitting "opioid overuse", to prescribe suboptimal doses of opioids to patients in need of them, and to prescribe less effective drugs to patients that are not similarly scheduled. This phenomemnon is described in the literature as "opiophobia" or "narcophobia". Scheduling of cannabinoid agonist would likely also have the unintentional consequence of causing physicians, fearful of being accused of permitting "overuse", to prescribe suboptimal doses of the drugs to patients in need of them, and to prescribe less effective drugs to patients that are not similarly scheduled. We have coined this phenomenon as "cannabinophobia" or "cannabiphobia".
[0025] There is a growing recognition in the medical community that a large number of patients suffer from the undertreatment of pain. Among the reasons frequently cited as causative of undertreatment are: (1) the failure to prescribe enough drug at the right dosage interval to reach a steady-state threshold
commensurate with the pain relief needed; (2) failure of patients to comply with a given dosage regimen; and (3) the reluctance of many physicians to prescribe analgesics categorized as controlled drugs based on often unfounded concerns of future addiction and fear of regulatory sanctions. For example, it has been reported that with respect to cancer pain, a large percentage of cancer patients suffer debilitating pain despite treatment with analgesics (Cleeland et al., New England Journal of Medicine 1994;330:592-596).
[0026] Attempts have been made to deter or minimize the abuse of orally administered opioids. These attempts have generally focused on the inclusion in the oral dosage form of an opioid antagonist, which is not orally active, but which will substantially block the analgesic effects of the opioid if one attempts to dissolve the opioid and administer it parenterally. A further evolution of this strategy has involved the inclusion in the oral dosage form of a sequestered, orally bioavailable opioid antagonist, which is released only upon product tampering (e.g., crushing, extraction). In this circumstance, the opioid antagonist is not expected to be orally active under normal conditions of use but would nullify the euphoriant effects of either oral or intravenous administration upon product tampering. Other attempts to deter or minimize the abuse of orally administered opioids have involved extended release formulations that are relatively resistant to mechanical (e.g., crushing), thermal (heating or melting) or chemical (e.g., alcohol) extraction and further, difficult to inject. This makes ingestion of the contents by an unintended route (e.g., intranasal, inhalation, intravenously or orally as a "bolus" in immediate release form) difficult. (See Sloan and Babul, Expert Opinion on Drug Delivery 2006, for recent review).
[0027] For example, commercially available Talwin™Nx tablets from Sanofi-
Winthrop contain a combination of pentazocine and naloxone. Pentazocine is a partial agonist at the μ opioid receptors and also has affinity at K opioid receptors, whereas, naloxone is an antagonist of μ receptors. Talwin Nx contains pentazocine hydrochloride equivalent to 50 mg base and naloxone hydrochloride equivalent to 0.5 mg base. Talwin Nx is indicated for the relief of moderate to severe pain. The amount of naloxone present in this combination
has no action when taken orally, and will not interfere with the pharmacologic action of pentazocine. However, this amount of naloxone given by injection has profound antagonistic action to opioid analgesics. Thus, the inclusion of naloxone is intended to curb a form of misuse of oral pentazocine, which occurs when the dosage form is solubilized and injected. Therefore, this dosage has lower potential for parenteral misuse than previous oral pentazocine formulations. Similarly, a drug known as Valoron™N (Goedecke), that comprises tilidine (50 mg) and naloxone (4 mg), has been available in Germany for the management of severe pain.
[0028] A fixed combination of buprenorphine and naloxone was introduced in
1991 in New Zealand (Temgesic™Nx, Reckitt & Colman) for the treatment of pain.
[0029] U.S. Patent No. 4,457,933 to Gordon et al. teaches the reduction in the oral abuse potential of the analgesics oxycodone, propoxyphene and pentazocine by combining the analgesic with naloxone in a specific range. Naloxone is combined with the selected analgesic a ratio of 2.5-5: 1 part.
[0030] U.S. Patent No. 6,228,863 to Palermo et al. teaches the reduction of the abuse potential of oral dosage forms of opioid analgesics by selecting the particular opioid agonist and antagonist pair, and the concentrations of the same such that the antagonist cannot be easily extracted from the agonist (at least a two-step extraction process being needed to separate the drugs-see also, WO 99/32120). The antagonist is in such a concentration that the combination will cause an aversive effect in a physically dependent human subject but not in a naive individual (See also, WO 99/32119).
[0031] U.S. Patent No. 3,773,955 to Pachter et al. describes orally effective analgesic compositions which contain from about 0.1 mg to about 10 mg naloxone with the opioid analgesic. Upon extraction of the composition, parenteral administration is dissuaded, as the dose of naloxone is high enough to prevent the production of analgesia, euphoria or physical dependence from the opioid analgesic. WO 01/58447 describes a controlled-release composition which contains an opioid agonist and opioid antagonist that provides an analgesic amount of the opioid agonist over 8 hours along with an amount of
opioid antagonist to attenuate a side effect of the opioid agonist. WO 01/58451 discloses an oral dosage form comprising an opioid agonist in releasable form and a sequestered opioid antagonist which is substantially not released when the dosage form is administered intact but is released upon tampering. As indicated above WO 99/32120 further describes selecting the opioid agonist and antagonist with respect to physical properties so as to require at least a two-step extraction process to separate the opioid agonist from the antagonist, the amount of opioid antagonist being otherwise sufficient to counteract opioid agonist effect if administered parenterally.
[0032] U.S. Patent No. 3,493,657 to Lewenstein, et al. describes compositions comprising naloxone and morphine or oxymorphone, which compositions were said to provide a strong analgesic effect without the occurrence of undesired side effects such as hallucinations.
[0033] U.S. Patent No. 4,582,835 to Lewis describes a method of treating pain by administering a sublingually effective dose of buprenorphine with naloxone. Lewis describes dosage ratios of naloxone to buprenorphine from 1 :3 to 1:1 for parenteral administration, and from 1:2 to 4: 1 for sublingual administration.
[0034] U.S. Patent No. 6,559,159 to Carroll et al. describes the use of kappa receptors antagonist for the treatment of opioid related addictions. One such compound is naltrexone, which is commercially available in the tablet form Revia™ for the treatment of alcohol dependence and for the blockade of exogenously administered opioids.
[0035] U.S. Patent Nos. 6,277,384, 6,375,957 and 6,475,494 describe oral dosage forms including a combination of an orally active opioid agonist and an orally active opioid antagonist in a ratio that, when delivered orally, is analgesically effective but that is aversive in a physically dependent subject.
[0036] - U.S. Patent Nos. 3,980,766, 4,070,494 and 6,309,668 describe formulations designed to prevent the injection of compositions meant for oral administration.
[0037] U.S. Patent No. 3,980,766 describes the incorporation of an ingestible solid which causes a rapid increase in viscosity upon concentration of an aqueous solution thereof.
[0038] U.S. Patent No. 4,070,494 describes the incorporation of a non-toxic, water gelable material in an amount sufficient to render the drug resistant to aqueous extraction.
[0039] U.S. Patent No. 6,309,668 describes a tablet for oral administration containing two or more layers comprising one or more drugs and one or more gelling agents within separate layers of the tablet. The resulting tablet forms a gel when combined with the volume of water necessary to dissolve the drug; this formulation thus reduces the extractability of the drug from the tablet. It should be noted that although these compositions preclude abuse by injection, this approach fails to prevent abuse by crushing and swallowing or snorting the formulation, which are commonly reported methods of abuse associated with OxyContin™.
[0040] U.S. Patent Nos. 3,773,955 and 3,966,940 describe formulations containing a combination of opioid agonists and antagonists, in which the antagonist does not block the therapeutic effect when the admixture is administered orally, but which does not produce analgesia, euphoria or physical dependence when administered parenterally by an abuser.
[0041] U.S. Patent No. 4,457,933 describes a method for decreasing both the oral and parenteral abuse potential of strong analgesic agents by combining an analgesic dose of the analgesic agent with an antagonist in specific, relatively narrow ratios.
[0042] The problem with all of the above schemes that incorporate opioid antagonists into the opioid preparation to deter abuse is that opioid antagonists themselves have side effects that may be disadvantageous. For example, nalorphine causes unpleasant reactions that range from anxiety, to "crazy feelings," to hallucinations, respiratory depression and miosis. Seizures have been reported with naloxone, albeit infrequently, and in postoperative patients, pulmonary edema and ventricular fibrillation have been seen with high dosages. Naltrexone has been reported to have the capacity to cause
hepatocellular injury when given in doses as low as fivefold or less of therapeutic doses. Nalmefene, although usually well tolerated, has been reported to cause nausea, vomiting and tachycardia in some individuals. Small doses of any of these opioid antagonists can also precipitate an abstinence syndrome in opioid tolerant patients, resulting in drug withdrawal. Symptoms of opioid withdrawal include body aches, diarrhea, gooseflesh, loss of appetite, nervousness or restlessness, runny nose, sneezing, tremors or shivering, stomach cramps, nausea, trouble with sleeping, increased sweating, increased yawning, weakness, increased heart rate or fever. These symptoms can be severe, requiring hospitalization and reinstitution of the opioid agonist.
[0043] Purdue Pharma (Euro-Celtique SA) have reported that one opioid tolerant volunteer among a 24-subject group receiving their extended release opioid agonist with a sequestered opioid antagonist developed severe opioid withdrawal, requiring hospitalization (Sloan and Babul, Expert Opinion on Drug Delivery 2006).
(0044] There is a need, therefore, for novel methods of deterring or preventing opioid abuse which do not require the incorporation of opioid antagonists into the formulation.
[0045] To date, no extended release formulations of opioids with abuse deterrent technology of any kind have; i) completed Phase III clinical trials; ii) been submitted for Marketing Application (New Drug Application) or iii) been commercialized anywhere in the world. Indeed if prior drug development history is any guide, most such strategies are unlikely to be developed or commercialized and the optimal formulation(s) will likely be apparent only through postmarketing surveillance of several formulations with competing technologies. In addition, regional differences in patterns of abuse means that different abuse deterrence strategies may be useful in different part of the world. Finally, experience with substance abuses indicates that those who are habitual abusers, particularly those who inject drugs intravenously, have a remarkable ability to defeat abuse deterrence strategies through physical and chemical manipulation of opioids and other drugs of abuse. Such addicts are frequently only one step behind strategies to deter abuse. With the ready
access to information from their well knit network and more recently, from websites on how to optimally extract the active agent from pharmaceutical dosage forms and maximize euphoriant effects, the development of abuse deterrent formulations has become a major pharmaceutical, clinical, regulatory and law enforcement challenge.
[0046] In view of this, it is not surprising that the Food and Drug
Administration's Division of Anesthetic, Analgesic and Rheumatology Drug Products and the U.S. Drug Enforcement Administration have encouraged companies to develop wide ranging abuse deterrent strategies for opioids, particularly extended release opioids and as "inducement", offered that such products may include in their prescribing information data about their products abuse deterrent properties (FDA Perspectives on Opioid Risk Management. Opioid Risk Management Meeting, Tufts Healthcare Institute, Boston, March 29, 2005; DEA Perspectives on Opioid Risk Management. Opioid Risk Management Meeting, Tufts Healthcare Institute, Boston, March 29, 2005).
[0047] In summary, various attempts have been made and are described in prior art to develop abuse-deterrent dosage forms. Clearly there is a need for a delivery system for opioid analgesics for patients seeking drug therapy and which deters abuse and minimizes or reduces the potential for psychological dependence.
[0048] One novel approach embodied by the present invention involves exploiting the interaction beteeen the cananbinoid and opioidergic systems in initiating and maintaining absue to mood altering systems.
[0049] Another novel approach embodied by the present invention involves modulating the abuse of opioid agonists by exploiting the direct and indirect role of cannabinoid antagonists in suppressing or nullyfing the mood altering effects of the opioid agonist.
[0050] Another novel approach embodied by the present invention involves deterring the abuse of the oral dosage form of the invention by targeting other abusable drugs which are not part of the invention, but which are frequently used, abused or co-abused with opioid agonists (e.g., cannabinoid agonist or alcohol).
[0051] The present invention involves oral dosage forms comprising an opioid agonist and an aversive agent chosen from among cannabinoid antagonists and drugs that produce aversion to alcohol, said aversive agents substantially non- releasable when the dosage form is taken intact and said dosage releasable upon tampering, said dosage form upon tampering effective in reducing or preventing drug abuse, drug diversion, mood alterations desired by drug abusers, said dosage form upon tampering also capable of producing adverse physiological and psychic conseqences.
DETAILED DESCRIPTION OF THE INVENTION
[0052] It is an object of the invention to provide an oral dosage form of an opioid agonist that is useful for decreasing the potential abuse of the opioid agonist without affecting the therapeutic effects of the opioid agonist or incurring the risk of precipitating signs and symptoms of opioid agonist withdrawal.
[0053] It is an object of the invention to provide an oral dosage form containing an effective dose of an opioid agonist along with a dose of cannabinoid antagonist which does not change the therapeutic efficacy of the opioid agonist when the dosage form is orally administered intact, but which can prevent abuse if the dosage form is tampered with by interfering with the effect of the opioid agonist.
[0054] It is an object of the invention to provide a method for preventing abuse of a oral opioid agonist dosage form where the dosage form also includes a dose of cannabinoid antagonist which is sequestered, e.g., is substantially not bioavailable when the dose is administered intact but is bioavailable when the dosage form is tampered with (e.g., in an attempt to misuse the dose of opioid agonist).
[0055] It is an object of the invention to provide oral pharmaceutical dosage forms that are intended for or are suitable for use in the management of acute or chronic diseases or disorders where alteration of the opioid agonist's
therapeutic effects must be avoided, as in cases of tolerance, physical dependence or individual variability in hepatic metabolism or physiology.
[0056] It is an object of the invention to provide a method of treating diseases or disorders in human subjects with an oral dosage form of an opioid agonist while reducing its misuse by oral, parenteral, intranasal, inhalational and/or sublingual route.
[0057] It is another object of the present invention to provide oral pharmaceutical compositions of an opioid agonist for decreasing the potential for abuse of the opioid agonist contained therein.
[0058] It is a further object of the invention is directed to provide oral pharmaceutical compositions of an opioid agonist which decrease the potential for abuse and co-abuse of cannabinoid agonists that are not part of the dosage form.
[0059] It is a further object of the present invention to provide oral pharmaceutical compositions of an opioid agonist for decreasing the potential for abuse and co-abuse of cannabinoid agonists in the setting of polydrug abuse.
[0060] It is a further object of the present invention to provide oral pharmaceutical compositions of an opioid agonist for indirectly deterring cannabinoid agonists abuse by drug addicts and/or recreational drug users by providing an aversive effect solely to the co-abused cannabinoid agonists in the setting of polydrug abuse.
[0061] It is a further object of the present invention to provide oral pharmaceutical compositions of an opioid agonist for preventing or minimizing the risk of opioid agonist toxicity from either intentional or unintentional tampering.
[0062] It is another object of the present invention to provide oral pharmaceutical compositions of an opioid agonist for deterring opioid agonist abuse by drug addicts and/or recreational drug users.
[0063] It is another object of the invention to provide oral pharmaceutical compositions of an opioid agonist which deters abuse by antagonizing co- abused substances that are not part of the dosage form, such as alcohol.
[0064] Some or all of the above objects and others are achieved by embodiments of the present invention, which is directed in part to an oral dosage form comprising an opioid agonist and an aversive agent which is present in a substantially non-releasable form (i.e., "sequestered"), said sequestered aversive agent comprising one or more cannabinoid antagonists, one or more alcohol deterrents or a combination thereof. In this way, the opioid agonists of the present invention can be formulated with a substantially non-releasable aversive agent to deter abuse and/or minimize opioid agonist toxicity on tampering.
[0065] In some preferred embodiments, the dosage form contains an orally therapeutically effective amount of the opioid agonist, the dosage form providing a desired analgesic effect. Because the cannabinoid antagonist is present in a substantially non-releasable form, it does not substantially block the therapeutic effects of the opioid agonist when the dosage form is orally administered intact, and does not pose a risk of precipitation of withdrawal in opioid tolerant or dependent patients.
[0066] One novel aspect of the invention concerns deterring or minimizing opioid agonist misuse, abuse and tampering by targeting other co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
[0067] Thus, in certain embodiments, the invention is directed in part to an oral dosage form comprising: (a) an opioid agonist; (b) an aversive agent which is sequestered in the intact dosage form but being releasable upon tampering of said dosage form, such that the intact dosage form releases 49% or less, preferably 42% or less, and more preferably 36% or less, 24.6% or less, 10% or less, or 6.2% or less of the aversive agent after 36 hours based on the in-vitro dissolution of. the dosage form in 900 ml of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm and 370C with a switch to Simulated Intestinal Fluid at 1 hour, the aversive agent when released upon tampering of said dosage form at least partially blocking the effect of the opioid agonist and/or at least partially blocking the effect of another abusable drug not included in the dosage form. Preferably, the intact
dosage form releases 42% or less, and more preferably 36% or less, 24.6% or less, 10% or less, or 6.2% or less of the aversive agent after 36 hours based on the in-vitro dissolution of the dosage form in 900 ml of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm and 37°C with a switch to Simulated Intestinal Fluid at 1 hour.
[0068] In certain embodiments, the invention is directed in part to an oral dosage form comprising (a) an opioid agonist in releasable form; (b) an aversive agent in substantially non-releasable form from the intact dosage form but being releasable upon tampering of said dosage form, such that the ratio of the mean Cmax of the aversive agent after single dose oral administration of the dosage form after tampering to the mean Cmax of aversive agent after single dose oral administration of an intact dosage form is at least 1.5: 1. Preferably, said ratio is at least 3: 1 ; or at least 6: 1; or at least 10: 1; or at least 20: 1; or at least 30: 1; or at least 40: 1; or at least 50: 1; or at least 70: 1; or at least 100:1; or at least 500:1.
[0069] In some preferred embodiments, the oral dosage form of the present invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers.
[0070] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4:1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated
Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the aversive agent is in the form of multiparticulates individually coated with a sequestering material which substantially prevents release of the aversive agent.
[0071] In another embodiment, the oral dosage form of the present invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in- vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the agonist and aversive agent are isolated from each other in two or more distinct layers.
[0072] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4.1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the aversive agent is dispersed in a matrix comprising a sequestering material which substantially prevents the release of the aversive agent.
[0073] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 1 hour is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of
Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers.
[0074] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 2 hour is about 4: 1 or greater, based on the in-vitro dissolution at 2 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
[0075] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 3 hour is about 4:1 or greater, based on the in-vitro dissolution at 3 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
[0076] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 4 hour is about 4: 1 or greater, based on the in-vitro dissolution at 4 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
[0077] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a
sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 6 hour is about 4:1 or greater, based on the in-vitro dissolution at 6 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
[0078] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of antagonist contained in the intact dosage form to the amount of the antagonist released from the intact dosage form after 8 hour is about 4: 1 or greater, based on the in-vitro dissolution at 8 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
[0079] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of antagonist contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 12 hour is about 4:1 or greater, based on the in-vitro dissolution at 12 hours of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
[0080] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 24 hour is about 4: 1 or greater, based on the in-vitro dissolution at 24 hours of the dosage form in 900 mL of
Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpra at 37 degrees C.
[0081] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent, wherein the agonist and aversive agent are interdispersed and visually indistinguishable.
[0082] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent, wherein the agonist and aversive agent are interdispersed and indistinguishable on the basis of physical characteristics, including bead diameter, density, texture, smell or flotation.
[0083] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) a therapeutic effect of an opioid agonist; and (ii) a sequestered aversive agent, such that at 1 hour after oral administration, the intact dosage form releases not more than about 25% of the aversive agent, the dosage form providing the therapeutic effects of the opioid agonist and the released aversive agent not affecting the therapeutic effects of the agonist, wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers. Preferably, the intact dosage form releases not more than about 12.5% of the aversive agent.
[0084] In other preferred embodiments, the invention is directed to an oral dosage form comprising: (i) an intended therapeutic effects of the opioid agonist in a releasable form; and an (ii) aversive agent in substantially non- releasable form wherein the aversive agent is in the form of multiparticulates individually coated with a material which substantially prevents release of the aversive agent.
[0085] In other preferred embodiments, the invention is directed to an oral dosage form comprising: (i) an opioid agonist in a releasable form; and a (ii) aversive agent in substantially non-releasable form wherein the aversive agent is dispersed in a matrix comprising a material which substantially prevents the release of the antagonist.
[0086] In other preferred embodiments, the invention is directed to an oral dosage form comprising an opioid agonist; and an aversive agent, in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
[0087] In other preferred embodiments, the invention is directed to an oral dosage form comprising an opioid agonist; and an orally-bioavailable aversive agent in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
[0088] In other preferred embodiments, the invention is directed to an oral dosage form comprising an opioid agonist; and an orally non-bioavailable aversive agent in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
[0089] In other preferred embodiments, the invention is directed to an oral dosage form comprising an opioid agonist; and aversive agent with low oral bioavailability in a substantially non-releasable form; wherein the agonist and aversive agent are at least partially interdispersed.
[0090] In embodiments of the invention wherein the aversive agent is in the form of multiparticulates coated with a sequestering material, the multiparticulates can be in the form of inert beads coated with the aversive agent and overcoated with a sequestering material, or alternatively in the form of a granulation comprising the antagonist and the material. The multiparticulates can be dispersed in a matrix comprising the opioid agonist or contained in a capsule with the opioid agonist.
[0091] In embodiments of the invention wherein the aversive agent is dispersed in a matrix comprising a sequestering material which substantially prevents the release of the aversive agent, the matrix can be in the form of pellets. The pellets can be dispersed in another matrix comprising the opioid agonist or contained in a capsule with the opioid agonist. The pellets can be visually distinct or visually indistinguishable.
[0092] In other preferred embodiments of the invention, a portion of the aversive agent is in a matrix and/or part of the aversive agent is in a coated bead.
[0093] In other preferred embodiments, the invention is directed to an oral dosage form comprising (i) an opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. wherein the aversive agent is in the form of multiparticulates individually coated with a sequestering material which substantially prevents release of the aversive agent and is subsequently overcoated with the opioid agonist such that each multiparticulate contains an aversive agent and an opioid agonist. In embodiments of the invention wherein the aversive agent is in the form of multiparticulates coated with a sequestering material, the multiparticulates can be in the form of inert beads coated with the aversive agent and overcoated with the sequestering material, and the overcoated still by the opioid agonist, or alternatively in the form of a granulation comprising the antagonist and the sequestering material, and the overcoated still by the opioid agonist. The multiparticulates can be dispersed in a matrix and compressed into a tablet or contained in a capsule.
[0094] In certain embodiments of the invention which exhibit the above- disclosed ratio of about 4: 1 or greater concerning the amount of aversive agent released from the dosage form after tampering to the amount of said aversive agent released from the intact dosage form based on the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C, the intact dosage form releases 22.5% or less of the aversive agent after 1 hour and the tampered dosage form releases 90% or more antagonist after 1 hour.
[0095] In another embodiment, the intact dosage form releases 20% or less of the aversive agent after 1 hour and the tampered dosage form releases 80% or more aversive agent after 1 hour.
[0096J In another embodiment, the intact dosage form releases 10% or less of said aversive agent after 1 hour and the tampered dosage form releases 40% or more aversive agent after 1 hour.
[0097] In another embodiment the intact dosage form releases 5% or less of said aversive agent after 1 hour and the tampered dosage form releases 20% or more aversive agent after 1 hour.
[0098] In certain embodiments of the invention, the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of said aversive agent released from the intact dosage form based on the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. is 10: 1 or greater, 50: 1 or greater or 100: 1 or greater.
[0099] In certain embodiments of the invention, the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of said aversive agent released from the intact dosage form based on the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. is 7.5: 1 or greater, 15: 1 or greater, 30: 1 or greater or 60:1 or greater.
[00100] In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 15% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 8% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 3% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non- releasable form is adapted to release less than 1% by weight in vivo after 36 hours. In certain embodiments of the dosage form the aversive agent in a substantially non-releasable form is adapted to release less than 0.5% by weight in vivo after 36 hours.
[00101] The invention is also directed to methods of preventing abuse of an opioid agonist utilizing the dosage forms disclosed herein. The invention is
also directed to methods of preventing abuse of a cannabinoid agonist utilizing the dosage forms disclosed herein, said agonist not part of the dosage form, said agonist used concurrently or contemporaneously with the dosage form. The invention is also directed to methods of preventing abuse of alcohol utilizing the dosage forms disclosed herein, said alcohol not part of the dosage form, said alcohol used concurrently or contemporaneously with the dosage form. The invention is also directed to methods of preventing abuse of a opioid agonists, cannabinoid agonists and/or alcohol in the setting of polydrug abuse or poly-substance abuse utilizing the dosage forms disclosed herein. The method for the foregoing can comprise providing the opioid agonist in an oral dosage form together with an aversive agent, wherein the aversive agent is present in a form which is substantially non-releasable form oral ingestion when the integrity of the dosage form is maintained at the time of oral ingestion, but which becomes bioavailable or releasable if subjected to tampering (e.g., crushing, grinding, pulverizing, heating, solvent immersion, solvent extraction, followed by oral, parenteral, intranasal or inhalational use or abuse).
[00102] Another embodiment of the invention is directed to a method of decreasing the abuse of an opioid agonist in an oral dosage form, comprising preparing an oral dosage form as disclosed herein. For example, the method can comprise preparing a dosage form which comprises (i) an orally therapeutically effective amount of an opioid agonist and (ii) an aversive agent in a substantially non-releasable form such that said dosage form provides a desired therapeutic effect and said antagonist does not substantially block the effect of the aversive agent when said dosage form is administered orally intact. In alternative embodiments, the effect of the opioid agonist is at least partially blocked when said dosage form is tampered with(e.g., crushing, grinding, pulverizing, heating, solvent immersion, solvent extraction, followed by oral, parenteral, intranasal or inhalational use or abuse).
[00103] The invention is also directed to a method of treating or preventing diseases and disorders amenable to treatment with opioid agonists with the dosage forms disclosed herein. The method can comprise providing an oral
dosage form containing an opioid agonist in a releasable form and an aversive agent in substantially non-releasable form; and orally administering the intact oral dosage form.
[00104] Another embodiment of the invention is directed to a method of preventing or treating pain with the disclosed dosage forms. In certain embodiments, the method of treating pain in patients with a dosage form having less abuse potential comprises providing an oral dosage form containing a releasable form of an opioid agonist and a substantially non- releasable form of an aversive agent; and orally administering the oral dosage form to provide a blood plasma level of agonist greater than the minimum analgesic concentration of the opioid agonist.
[00105] Another embodiment of the invention is directed to a method of preventing or treating diseases and disorders amenable to treatment with opioid agonists with the disclosed dosage forms. In certain embodiments, the method of preventing or treating such diseases and disorders in patients with a dosage form having less abuse potential comprises providing an oral dosage form containing a releasable form of an opioid agonist and a substantially non- releasable form of an aversive agent; and orally administering the oral dosage form to provide a blood plasma level of agonist greater than the minimum analgesic concentration of the opioid agonist.
[00106] The invention is also directed to methods of preparing the dosage forms disclosed herein. In certain embodiments, the invention comprises a method of preparing an oral dosage form comprising pretreating an aversive agent to render it substantially non-releasable; and combining the pretreated aversive agent with a releasable form of opioid agonist in a manner that maintains the integrity of the non-releasable form of the aversive agent.
[00107] In some preferred embodiments of the invention, the one or more aversive agents in sequestered (i.e., non-releasable or substantially releasable) form are chosen from the group comprising cannabinoid antagonists and alcohol deterrents.
[00108] Certain embodiments of the invention are directed to formulations wherein the agonist and aversive agent are interdispersed and are not isolated
from each other in two distinct layers. However in certain embodiments, the agonist and aversive agent are partially interdispersed.
[00109] In certain preferred embodiments of the invention, the dosage form comprises one or more opioid agonists in releasable or substantially releasable form; and one or more aversive agents in a non-releasable or substantially releasable form when said dosage form is used as intended.
[00110] In certain preferred embodiments of the invention, the dosage form comprises one or more opioid agonists in releasable or substantially releasable form, and one or more cannabinoid antagonists and one or more opioid antagonists, each in a non-releasable or substantially releasable form, when said dosage form is used as intended.
[00111] In certain preferred embodiments of the invention, the dosage form comprises one or more opioid agonists in releasable or substantially releasable form, and one or more cannabinoid antagonists plus one or more alcohol deterrents, each in a non-releasable or substantially releasable form, when said dosage form is used as intended.
[00112] In certain preferred embodiments of the invention, the dosage form comprises one or more opioid agonists in releasable or substantially releasable form, and one or more opioid antagonist plus one or more alcohol deterrents, each in a non-releasable or substantially releasable form, when said dosage form is used as intended.
Blunting the Effects of Opioid Agonists, Co-abused Cannabinoid Agonists and Alcohol
[00113] Mammalian tissues express at least two cannabinoid receptors, both of which are G-protein coupled. These are CBj receptors and CB2 receptors. CBj receptors are expressed are primarily expressed in peripheral and central nerve terminals where they mediate inhibition of neurotransmitter release. In the CNS, especially high levels of CBi receptors are found in the cerebellum, hippocampus and basal ganglia. CB2 receptors are found primarily on immune and hematopoietic cells outside (and also within) the central nervous system, where they appear to modulate cytokine release and immune cell migration. Studies using CBi and CB2 receptor knockout mice indicate that some of the
effects of endocannabinoids are not mediated by either CB i or CB2 receptors, suggesting the existence of additional yet to be identified sites of action. Some cannabinoid effects resist classification as either CB) and CB2-mediated. There is growing evidence suggesting the involvement of additional receptors, which include TRPVi receptors and at least 2 G protein-coupled receptors (GPCRs) of unclear molecular identity that have only been defined pharmacologically.
[00114] The human cannabinoid system is involved in a number of pathological states, including Alzheimer's disease, schizophrenia, depression, alcoholism, Parkinson's disease, stroke, premature labor, endotoxic shock, hepatic cirrhosis, atherosclerosis, cancer, bone implantation, glaucoma, emesis and pain. Additionally, upregulation or downregulation of the endocannabinoid system is seen in a variety of animal in vivo models, including multiple sclerosis, amyotrophic lateral sclerosis, encephalitis, Alzheimer's disease, Parkinson's disease, Huntington's disease, pain, obesity, feeding, fasting, stress, memory, aging, hypertension, cirrhosis, septic shock, cardiogenic shock, cerebral ischemia, myocardial infarction, neurotoxicity, febrile seizures and various intestinal disorders.
[00115] Therefore cannabinoid receptors present a large number of potential targets for pharmacologic intervention and efforts are underway to develop and test a variety of cannabinoid agonists and antagonists to prevent and treat various maladies. Presently, three non-specific cannabinoid receptor agonists are commercially available. Nabilone (Cesamet™) and dronabinol (Marinol™) are oral synthetic THC analogs which have been shown effective for the treatment of nausea and vomiting associated with cancer chemotherapy and AIDS-related cachexia. A buccal spray containing THC and cannabidiol (Sativex™) was approved in Canada for the symptomatic relief of neuropathic pain in multiple sclerosis.
[00116] All cannabinoid agonists are scheduled under the Controlled
Substances Act of 1970. Cannabinoid agonists can produce a variety of adverse effects including^ a number of psychotomimetic effects such as dizziness, drowsiness, euphoria, ataxia, anxiety, disorientation, depression,
hallucinations, vertigo, and psychosis. While these psychic effects are undesirable for patients, they are often sought after by recreational drug users and individuals with an addiction disorder. Cannabinoids play a modulatory role in drug seeking. They can reinstate cocaine seeking behavior after several weeks of extinction of intravenous cocaine self-administration. Similar effects have been shown in animals with a history of heroin, methamphetamine, alcohol and nicotine self-administration where cannabinoid receptor agonists have reinstated previously abolished drug seeking.
[00117] There is significant concern on the part of addiction medicine specialists and public health regulators about the potential risk of drug abuse and drug diversion with the commercialization and widespread use of new cannabinoid agonists currently in development.
[00118] Addiction to drugs is characterized by long-lasting motivational disturbances including compulsive drug seeking, intense drug craving, use despite harm, the non-medical use and diversion of psychoactive substances, manipulation of the medical system and escalating drug use and risk taking behaviors. The neurobiological mechanisms underlying such behaviors are poorly understood. Cannabinoids play a modulatory role in drug seeking. An early signal came from the observation that the potent cannabinoid receptor agonist (6aR)-trans-3-( 1 , 1 -dimethylheptyl)-6a,7, 10, 1 Oa-tetrahydro- 1 -hydroxy- 6,6-dimethyl-6H-dibenzo [b,d] pyran-9-methanol was able to reinstate cocaine seeking behavior after several weeks of extinction of intravenous cocaine self- administration in the rat (De Vries, Nat Med, 2001). Further, this effect was completely abolished by the selective CBi receptor antagonist N-piperidinyl- 5-(4-chlorophenyl)- 1 -(2,4-dichlorophenyl)-4-methyl pyrazole-3-carboxamide, suggesting a role for cannabinoid agonists in cocaine relapse.
[00119] Similar effects have been shown in animals with a history of heroin, methamphetamine, alcohol and nicotine self-administration where cannabinoid receptor agonists have reinstated previously abolished drug seeking (De Vries et al, Psychopharmacology [Berl], 2003; Fattore et al, Eur J Neurosci, 2003; De Vries et al., Behav Brain Res, 2005; Anggadiredja et al., Neuropsychopharmacology, 2004).
[00120] In vitro studies have shown that cannabinoid agonists and opioid agonists activate mu, delta and kappa opioid, and CB], CB2 and non-CB]/CB2 cannabinoid receptors, respectively, which are coupled to Gi/Go GTP-binding proteins that inhibit adenylyl cyclase, inhibit voltage-dependent calcium channels, stimulate potassium channels and activate the MAP kinase cascade (for review see Childers, 1991; Childers et al., 1992; Howlett, 1995).
[00121] Receptor mapping studies have shown a rather similar distribution of
CB i cannabinoid and μ-opioid receptors in the dorsal horn of the spinal cord (Welch and Stevens 1992; Hohmann et al., 1999; Salio et al., 2001) and in the CNS, including the caudate, putamen, dorsal hippocampus, and substantia nigra (Mansour et al., 1988; Herkenham et al., 1991; Mailleux and Vanderhaeghen, 1992; Rodriguez et al., 2001).
[00122] Chronic use of cannabinoid agonists and opioid agonists results in pharmacologic tolerance, physical dependence and addiction. Chronic cannabinoid agonist administration induces tolerance to the antinociceptive effect of opioids (Smith et al., 1994; Welch, 1997), while chronic exposure to opioid agonists results in tolerance to the antinociceptive effect of cannabinoid agonists (Bloom and Dewey, 1978; Hine, 1985; Smith et al., 1994; Thorat and Bhargava, 1994). Cross-physical dependence between opioid agonists and cannabinoid agonists has also been demonstrated (Bhargava, 1976, 1978; Hine et al., 1975; Vela et al., 1995; Yamaguchi et al., 2001; Del Arco et al., 2002). Administration of the opioid antagonist naloxone precipitates an abstinence syndrome in cannabinoid-tolerant rats (Hirschhorn and Rosecrans, 1974; Kaymakcalan et al., 1977). Similarly, the cannabinoid antagonist SR141716A precipitates abstinence in opioid agonist dependent rats (Navarro et al., 1998). Sustained suppression of CBi receptor activity with the cannabinoid antagonist SR141716A during opioid agonist administration reduces the signs and symptoms of opioid withdrawal (Rubino et al., 2000; Mas-Nieto et al., 2001).
[00123] Cannabinoid agonists and opioid agonists seem to interact in their antinociceptive effects as illustrated by the ability of their respective antagonists to reverse cannabinoid/opioid-induced analgesia (Welch, 1993;
Reche et al., 1996a,b; Cichewicz et al., 1999). The concurrent administration of opioid agonsits and cannabinoid agonists results in an enhanced antinociceptive effect, compared with either solo administartion (Cichewicz et al., 1999; Smith et al., 1998; Welch and Eads, 1999; Cichewicz and McCarthy, 2003). Administration of subanalgesic and submaximal doses of cannabinoid agonists and opioid agonists result in synergy and this effect is abolished by cannabinoid receptor and opioid receptor antagonists (Reche et al., 1996a; Smith et al., 1998; Cichewicz, 2004).
[00124] The reward process is central to the development of addiction to psychoactive drugs. A commonly used experimental method of evaluating the reinforcing properties of drugs is the self-administration test. Available data suggest that there is an an interaction between opioids and cannabinoids with respect to reward processes. The cannabinoid antagonist SR141716A reduces self-administration of heroin (Chaperon et al., 1998; Braida et al., 2001; Mas- Nieto et al., 2001; Navarro et al., 2001; De Vries et al., 2003). The opioid antagonists naltrexone and naloxone reduce self-administration of THC (Tanda et al., 2000; Justinova et al., 2003, 2004) and the CBl agonist CP- 55,940 (Braida et al., 2001). Cannabinoid antagonists can also suppress "heroin-seeking" behavior after weeks of prior extinction (Fattore et al., 2003; Caille and Parsons, 2003; Solinas et al., 2003).
[00125] A majority of opioid-dependent individuals seeking treatment are polydrug abusers. The secondary licit or illicit abusable or mood altering drug used most frequently in this population is marijuana and alcohol. Prevalence estimates of marijuana use have ranged from 25% to 80% among cocaine and opioid agonist abusers (Ball et al., 1988; Budney et al., 1996;Miller et al., 1990; Nirenberg et al., 1996; Saxon et al.,1993). Budney et al (Addiction, 1998) evaluated marijuana use among opioid abusers in patients enrolled in treatment for opioid dependence. Sixty-six per cent of participants were current marijuana users and almost all (94%) continued to use during treatment. In another study (Budney et al., Drug Abuse and Dependence, 1996) examining the relationship between marijuana use and sociodemographic, psychosocial, and drug-use variables in treatment-seeking
opioid abusers, marijuana involvement was associated with less stable relationships, more frequent alcohol use, more financial difficulty, and engagement in more risky behavior including intravenous drug use and needle-sharing.
[00126] Forensic analytical toxicology studies have repeatedly revealed that in a majority of subjects, there are multiple drugs of abuse in the systemic circulation and importantly, such polydrug abuse is believed to be an important contributor to the death of many subjects from drug abuse and intentional or unintentional drug overdose. This observation of the use of multiple drugs of abuse in the same subject, i.e., polydrug abuse has also been reported in the drug abuse and addiction medicine literature, where positive urine samples ("dirty urine") frequently contains more than one illicit drug.
[00127] For example, a majority of opioid agonist users are polydrug abusers.
A frequently co-abused substance in this population is alcohol.
[00128] There is a need, therefore, for novel methods and compositions for deterring or preventing opioid agonist abuse by targeting the role of cannabinoid agonists in initiating and maintaining physical dependence, psychological dependence, tolerance and addiction to the opioid agonist.
[00129] Methods of abuse deterrence that make it difficult to tamper with the product by creating significant barriers to mechanical, thermal and solvent extraction prior to ingestion have their own challenges. In some cases, such formulations are difficult to manufacture and may not offer sufficient protection against tampering, since the drug must eventually be in releasable from in order to be delivered through the gastrointestinal tract.
[00130] There is also a need for novel methods and compositions for deterring or preventing opioid agonist abuse when said agonist is formulated as an extended release formulation.
[00131] One novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target the role of cannabinoid antagonist in nullifying the mood altering effects of opioid agonists.
[00132] Another novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target the role of cannabinoid antagonist in
producing an aversive effects and in precipitating ari abstinence syndrome (withdrawal syndrome) in subjects tolerant to opioid agonists.
[00133] Yet another novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target other co-consumed or co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
(00134] The observation of polydrug abuse can be exploited to deter opioid agonist abuse indirectly by antagonizing the pleasurable and mood altering effects of cannabinoid agonists and alcohol which are not part of the dosage form of the invention but may nevertheless be co-abused by opioid agonist abusers.
[00135] There are no described abuse or misuse deterrence methods for opioid agonists, involving coadministration with opioid antagonists such as rimonabant.
[00136] There are no described abuse or misuse deterrence methods for opioid agonists, including coadministration with alcohol deterrents such as disulfiram or calcium carbimide.
[00137] There is therefore a need for abuse and tamper deterrent, and generally safer dosage forms of opioid agonists which deter abuse by: (i) providing an aversive effect towards co-consumed or co-abused mood altering substances such as alcohol and cannabinoid agonists which are not part of the dosage form; (ii) nullifying the mood altering effects of the abused opioid agonist; and/or (iii) providing an aversive or adverse effect directed at the opioid agonist and any co-consumed opioid agonists or alcohol.
[00138] The present invention relates to oral opioid agonist in releasable form and aversive agents in substantially non-releasable form (i.e., sequestered), said dosage forms having reduced potential for abuse in opioid agonist abusers and in polydrug abusers. Under conditions of abuse or tampering (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered aversive agent (e.g., alcohol deterrent or cannabinoid antagonist) on: (i) co-used or co-abused
alcohol or cannabinoid agonist, particularly in the setting of polydrug abuse and (ii) nullification of the mood altering effects of the opioid agonist.
[00139] The present invention is directed to immediate and controlled release oral dosage form of opioid agonists which, are formulated in order to reduce and minimize misuse, abuse and diversion. In certain embodiments, these characteristics are conferred by the inclusion of a cannabinoid antagonist, which is itself formulated in a unique controlled release matrix. The properties of this formulation are developed to liberate the cannabinoid antagonist in conditions of misuse or tampering yet a negligible amount of cannabinoid antagonist would be released (an amount which does not affect therapeutic effect of the opioid agonist experienced by the patient) under the prescribed conditions of use.
[00140] The present invention is based in part on several observations: (i) opioid agonists (or opioid analgesics) are frequently abused in the setting of polydrug abuse (e.g., co-abuse with marijuana or alcohol) and (ii) the endocannabinoid system interacts with the opioidergic system. These observations can be exploited to deter opioid agonist abuse by including in the opioid agonist dosage form a substantially non-releasable cannabinoid antagonist in quantities sufficient to nullify the effects of the abused opioid agonist and/or the co-used or co-abused cannabinoid agonist, said cannabinoid agonist not part of the dosage form. These observations can also be exploited to deter opioid agonist abuse by including in the opioid agonist dosage form a substantially non-releasable alcohol deterrent in quantities sufficient to produce and aversive or adverse effects on any co-used or co-abused alcohol, said alcohol not part of the dosage form.
[00141] In certain embodiments, the invention provides a method for preventing abuse of a oral opioid agonist dosage form where the dosage form also includes a dose of cannabinoid antagonist or alcohol deterrent which is sequestered, e.g., is substantially not bioavailable when the dose is administered intact but is bioavailable when the dosage form is tampered with (e.g., in an attempt to misuse the dose of opioid agonist).
[00142] In certain embodiments, the invention to provide an oral dosage form of an opioid agonist and a substantially non-releasable cannabinoid antagonist that is useful for decreasing the potential abuse of the opioid agonist when administered intact, without affecting the therapeutic effects of the opioid agonist or incurring the risk of precipitating signs and symptoms of opioid agonist withdrawal.
[00143] In some preferred embodiments, the sequestered cannabinoid agonist upon tampering becomes substantially releasable and produces its abuse and misuse deterrence by nullifying some or all of the mood altering effects of the cannabinoid agonist co-abused by the subject or by precipitating an abstinence syndrome (i.e., signs and symptoms of cannabinoid agonist withdrawal) in subjects with cannabinoid agonist tolerance and physical dependence.
[00144] Without being bound by theory, in some embodiments, the sequestered cannabinoid antagonists upon tampering releases some or a substantial amount of its cannabinoid antagonist content from the dosage form of the invention, thereby nullifying some or a substantial amount of the mood altering effect of the opioid agonist, as well as any co-abused cannabinoid agonist that subject has taken in the past, is taking concurrently or is expected to take in the near future (e.g., cannabinoid agonist use within minutes, hours or days of use of the tampered dosage form of the invention). In other embodiments, the sequestered cannabinoid antagonists upon tampering releases some or a substantial amount of its cannabinoid antagonist content from the dosage form of the invention, thereby precipitating an opioid agonist abstinence syndrome (i.e., signs and symptoms of opioid agonist withdrawal), as well as a cannabinoid agonist abstinence syndrome (i.e., signs and symptoms of cannabinoid agonist withdrawal), in subjects with opioid agonist and cannabinoid agonist tolerance and physical dependence, respectively.
[00145] In certain embodiments of the present invention, the ratio of the opioid agonist to the substantially non-releasable form of the aversive agent in the oral dosage form is such that the effect of the opioid agonist is at least partially blocked when the dosage form is chewed, crushed or dissolved in a solvent and heated, and administered orally, intranasally, inhalationally, parenterally
or sublingually. Since the oral dosage form of the present invention, when administered properly as intended, would not substantially release the cannabinoid antagonist, the amount of such antagonist may be varied more widely than if the cannabinoid antagonist is available to be released into the gastrointestinal system upon oral administration. For safety reasons, the amount of the antagonist present in a substantially non-releasable form should not be permanently harmful to humans even if fully released. The ratio of particular cannabinoid agonist to antagonist can be determined by one skilled in the art.
[00146] In certain embodiments of the present invention, the ratio of the opioid agonist to the aversive agent, present in a substantially non-releasable form, is about 1:10,000 to about 10000:1 by weight, or about 1 :1000 to about 1000: 1 by weight or preferably about 1 : 10 to about 10: 1 by weight, and more preferably about 5: 1 to 1:5 by weight. The weight ratio of the opioid agonist to aversive agent, as used in this application, refers to the weight of the active ingredients.
Definitions
[00147] The term "analgesic effectiveness" is defined for purposes of the present invention as a satisfactory prevention, reduction in or elimination of pain, along with a tolerable level of side effects, as determined by the human patient.
[00148] The term "therapeutic effectiveness" is defined for purposes of the present invention as a satisfactory prevention or treatment of diseases and disorders amenable to treatment with an opioid agonist, including their signs and symptoms, along with a tolerable level of side effects, as determined by the human patient.
[00149] An "agonist" is a ligand that binds to a rebeptor and alters the receptor state resulting in a biological response. Conventional agonists increase receptor activity, whereas inverse agonists reduce it (See Neubig et al, IUPHAR Committee on Receptor Nomenclature and Classification, Pharmacol Rev, 2003; Howlett et al., MoI Pharmacol, 1988).
[00150] An "antagonist" is a drug of ligand that reduces the action of another drug or ligand, generally an agonist. Many antagonists act at the same receptor macromolecule as the agonist. (See Neubig et al, IUPHAR Committee on Receptor Nomenclature and Classification, Pharmacol Rev, 2003; Howlett et al., MoI Pharmacol, 1988).
[00151] The term "receptor" means a molecule within a cell, on a cell surface, on a membrane, in tissue, in fluid or otherwise found in humans that serves as a recognition or binding site to cause specific physiologic, pathophysiologic or pharmacologic effects. The term "receptor" also means a cellular macromolecule, or an assembly of macromolecules, that is concerned directly and specifically in chemical signaling between and within cells. Combination of a hormone, neurotransmitter, drug, ligand, or intracellular messenger with its receptors) initiates a change in cell function (Neubig et al, IUPHAR Committee on Receptor Nomenclature and Classification, Pharmacol Rev, 2003).
[00152] The term "a cannabinoid antagonist in a substantially non-releasable form" , "a cannabinoid antagonist in a non-releasable form", "an alcohol deterrent in a substantially non-releasable form", "an alcohol deterrent in a non-releasable form", "an aversive agent in a substantially non-releasable form" or "an aversive agent in a non-releasable form", refer to said agent that is not released or substantially not released at one hour after the intact dosage form containing both opioid agonist and the said agent is orally administered (i.e., without having been tampered with). For purposes of the invention, the amount released after oral administration of the intact dosage form may be measured in-vitro via the dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C. Such a dosage form is also referred to as comprising a "sequestered aversive agent", or a "sequestered agent", or as applicable, a "sequestered cannabinoid antagonist", or as applicable, a "sequestered alcohol deterrent".
[00153] The term "tampering" or "tamper" means any manipulation including by mechanical, thermal and/or chemical means which changes the physical
properties of the dosage form, e.g., to liberate the opioid for immediate release if it is in sustained release form, or to make the opioid agonist available for inappropriate use such as administration by an alternate route, e.g., parenterally inhalationally, intranasally. The tampering can be, e.g., by means of crushing, shearing, grinding, chewing, dissolution in a solvent, heating, mechanical extraction, solvent extraction, solvent immersion, combustion, or any combination thereof.
[00154] The term "at least partially blocking the [e.g., cannabinoid agonist or opioid agonist or alcohol] effect" is defined for purposes of the present invention to mean that the cannabinoid antagonist at least partially or at least significantly blocks the mood altering effects of the opioid agonist, or the cannabinoid agonist or alcohol taken separately by the human subject or patient, thereby reducing the potential for abuse of the opioid agonist in the dosage form.
[00155] The term "mood altering" is defined for purposes of the present invention to mean that the "high", "liking", pleasurable, euphoric, calming, anxiolytic, auditory and visual perceptual alterations, relaxing, psychotomimetic, mood altering, rewarding, reinforcing alterations in perception, cognition and mental focus; sexual gratification; sexual arousal; sexual desire and sexual anticipation; increased socialization effects of the abusable drug.
[00156] The term "abuse", "misuse", "cannabinoid abuse", "cannabinoid agonist abuse", "cannabinoid agonist misuse", "opioid agonist abuse", "opioid agonist misuse" or "opioid abuse" in the context of the present invention, means single use, intermittent use, repeated use, recreational use and chronic use of the specified abusable drug or class of abusable drugs: (i) in quantities or by methods and routes of administration that do not conform to standard medical practice; (ii) outside the scope of specific instructions for use provided by a qualified medical professional; (iii) outside the supervision of a qualified medical professional; (iv) outside the approved instructions on proper use provided by the drug's legal manufacturer; (v) which is not in specifically approved dosage forms for medical use as pharmaceutical agents;
(vi) where there is an intense desire for and efforts to procure same; (vii) compulsive use; (viii) through acquisition by manipulation of the medical system, including falsification of medical history, symptom intensity, disease severity, patient identity, doctor shopping, prescription forgeries; (ix) where there is impaired control over use; (x) despite harm; (xi) by procurement from non-medical sources; (xii) by others through sale or diversion by the individual into the non-medical supply chain; (xiii) for medically unapproved or unintended mood altering purposes. The term "abuse resistant", "abuse deterrent", "tamper resistant",
"deter abuse" ", "deter misuse", resist abuse" and "resist misuse" and "deter abuse" (as well of the words "resist" or "deter" when applied to abusable drugs of the invention) are used interchangeably in the context of the present invention and include pharmaceutical compositions and methods that resist, deter, discourage, diminish, delay and/or frustrate: (i) the intentional, unintentional or accidental physical or chemical manipulation or tampering of the dosage form (e.g., crushing, shearing, grinding, chewing, dissolving, melting, needle aspiration, inhalation, insufflation, extraction by mechanical, thermal and chemical means, and/or filtration); (ii) the intentional, unintentional or accidental use or misuse of the dosage form outside the scope of specific instructions for use provided by a qualified medical professional, outside the supervision of a qualified medical professional and outside the approved instructions on proper use provided by the drug's legal manufacturer (e.g., intravenous use, intranasal use, inhalational use and oral ingestion to provide high peak concentrations); (iii) the intentional, unintentional or accidental conversion of an extended release dosage form of the invention into a more immediate release form; (iv) the intentional and iatrogenic increase in physical and psychic effects sought by recreational drug users, addicts, and patients with pain who have an addiction disorder; (v) attempts at surreptitious administration of the dosage form to a third party (e.g., in a beverage); (vi) attempts to procure the dosage form by manipulation of the medical system and from non-medical sources; (vii) the sale or diversion of the dosage form into the non-medical supply chain and for medically
unapproved or unintended mood altering purposes; (viii) the intentional, unintentional or accidental attempts at otherwise changing the physical, pharmaceutical, pharmacological and/or medical properties of the dosage form from what was intended by the manufacturer.
[00158] As used herein, the term "aversive agents", "aversion producing agents" and "aversive compounds" means to compounds contained within the dosage form that produce an aversive, undesirable, repugnant, distasteful, unpleasant, unacceptable physiologic or unacceptable psychic effects, or alcohol deterrence, or that pharmacologically block or reduce the mood altering effects of the dosage form or of concurrently or contemporaneously used abusable drugs, preferably selected from the group comprising cannabinoid agonists, alcohol of combinations thereof.
[00159] The term "sustained release" is defined for purposes of the present invention as the release of the cannabinoid agonist from the oral dosage form at such a rate that blood (e.g., plasma) concentrations (levels) are maintained within the therapeutic range (above the minimum effective concentration) but below toxic levels over a period of 4 to 24 hours, preferably over a period of time indicative of a rwice-a-day or a once-a-day formulation. As used herein, "sustained release" is interchangeable with "extended release", "controlled release", "modified release", "delayed release" and the like.
[00160] The term "subject" for purposes of treatment is used interchangeably with "patient", "male", "female", and includes any human who has a medical condition amenable to prevention or treatment with an opioid agonist.
[00161] The terms "medical condition", "malady", "disease", "disorder" and
"pathological states" are used interchangeably and are intended to have their broadest interpretation to refer to any physiologic, pathologic or pathophysiologic state in a human, including the signs and symptoms thereof that can be prevented, treated, managed or altered to produce a desired, usually beneficial effect.
[00162] "Drug", "drug substance", "substance", "therapeutic agent",
"pharmacological agent", "pharmaceutical agent", "active agent" and "agent" are used interchangeably and are intended to have their broadest interpretation
as to any therapeutically active substance which is delivered to a living organism to produce a desired, usually beneficial effect. In general, this includes therapeutic agents in all of the major therapeutic areas.
[00163] "Pharmaceutically or therapeutically acceptable excipient or carrier" refers to a solid or liquid filler, diluent or encapsulating substance which does not interfere with the effectiveness or the biological activity of the cannabinoid agonist and which is not toxic to the hosts, which may be either humans or animals, to which it is administered.
[00164] The term "pharmaceutically acceptable salt" as used herein refers to a salt which is toxicologically safe for human and animal administration. Nonlimiting examples of salts include hydrochlorides, hydrobromides, hydroiodides, sulfates, bisulfates, nitrates, citrates, tartrates, bitartrates, phosphates, malates, maleates, napsylates, fumarates, succinates, acetates, terephlhalates, pamoates and pectinates.
[00165] In some embodiments, the xenobiotic pharmaceutical composition is a salt or complex of inorganic cation salts, organic salts such- primary, secondary, tertiary and quaternary amines include substituted amines In some embodiments, examples of suitable pharmaceutically acceptable salts of xenobiotic include any of the inorganic cation salts such as sodium, potassium, lithium, magnesium, calcium, cesium, ammonia, ferrous, zinc, manganous, aluminum, ferric, and manganic; organic salts with primary, secondary, tertiary and quaternary amines, or mixtures thereof. Examples of such primary, secondary, tertiary and quaternary amines include substituted amines including but not limited to naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and mixtures thereof. More specifically, suitable amines include but are not limited to tromethamine, triethylamine, tripropylamine, dropopizine, 2-dimethylaminoethanol, 2- diethylaminoethanol, lysine, arginine, ornithine, histidine, caffeine, procaine, N-ethylpiperidine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, frø-(hydroxymethyl)aminomethane, Ν-methylglucamine, methylglycamine, theobromine, piperazine, piperidine, polyamine resins and the like, and mixtures thereof.
[00166] In some embodiments, examples of suitable pharmaceutically acceptable salts include aminoalcohols chosen from the group consisting of ethanolamine, 3-amino-l-propanol, (7?)-l-amino-2-propanol, (5)-l-amino- 2-propanol, 2 -amino- 1 ,3-propandiol, N-(2-hydroxyethyl)pyrrolidine, D-glucamine and L-prolinol, D-glucosamine, and N-methylglucosamine.
[00167] In some embodiments, examples of suitable pharmaceutically acceptable salts include alkali and alkaline earth metals and salts of an organic nature, such as the salts of basic amino acids.
[00168] Some of the opioid agonists, cannabinoid antagonists and alcohol deterrents disclosed herein may contain one or more asymmetric centers and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms. The present invention is also meant to encompass all such possible forms as well as their racemic and resolved forms and mixtures thereof. When the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended to include both E and Z geometric isomers. All tautomers are intended to be encompassed by the present invention as well.
[00169] As used herein, the term "stereoisomers" is a general term for all isomers of individual molecules that differ only in the orientation of their atoms is space. It includes enantiomers and isomers of compounds with more than one chiral center that are not mirror images of one another (diastereomers).
[00170] The term "chiral center" refers to a carbon atom to which four different groups are attached.
[00171] The term "enantiomer" or "enantiomeric" refers to a molecule that is nonsuperimposeable on its mirror image and hence optically active wherein the enantiomer rotates the plane of polarized light in one direction and its mirror image rotates the plane of polarized light in the opposite direction.
[00172] The term "racemic" refers to a mixture of equal parts of enantiomers and which is optically inactive.
[00173] The term "resolution" refers to the separation or concentration or depletion of one of the two enantiomeric forms of a molecule.
Aversive Agents
[00174] Aversive agents of the oral dosage form of the invention comprise one or more cannabinoid agonists, one or more alcohol deterrents or mixtures there of, said aversive agent(s) in the dosage form in substantially non-releasable form. When the dosage form is tampered, the aversive agent becomes partially or substantially releasable.
[00175] In some embodiments, the aversive agents are in the form of pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
[00176] In certain embodiments, the amount of substantially non-releasable aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists and alcohol deterrents may be about 10 ng to 1500 mg, more preferably, 10 ng to 1000 mg, even more preferably 0.1 mg to 800 mg, and most preferably, 0.1 mg to 500 mg.
[00177] The amount of aversive agent required to produce the desired aversive effect will vary, but can be readily determined based on the physicochemical and pharmaceutical properties and the pharmacology of the drug, including its oral bioavailability, half life, intrinsic clearance, potency, absolute bioavailability, potency, safety and the like, and the magnitude of desired aversive effect.
Cannabinoid Agonists and Cannabinoid Antagonists
[00178] The term "cannabinoid agonist" means a substance that binds to one or more cannabinoid receptor to exert an agonist or partial agonist effect.
[00179] The term "cannabinoid antagonist" means an antagonist substance or an inverse agonist that binds to one or more cannabinoid receptor to exert an antagonist effect.
[00180] The term "cannabinoid receptor" means a molecule that causes a specific physiologic, pathophysiologic or pharmacologic effect after binding to CBi, CB2, non-CBi/CB2 cannabinoid sites, TRPVi receptors, as well as
other G protein-coupled receptors (GPCRs) that form part of the endocannabinoid system (Wiley and Martin, Chemistry Physics of Lipids, 2002; Begg et al., Pharmacol Ther, 2005; Howlett et al., Neuropharmacol, 2004; Pertwee, AAPS Journal, 2005; International Union of Pharmacology (IUPHAR) Receptor Database; Howlett et al., MoI Pharmacol, 1988).
[00181] A number of assays are available to determine whether a drug is a cannabinoid agonist or cannabinoid antagonist, using in vivo and in vitro bioassay systems (Howlett et al., MoI Pharmacol, 1988).
[00182] Cannabinoid agonists are known or readily determined by individuals who practice the art. Preferably, the cannabinoid agonist useful for the present invention may be selected from the group consisting of inhibitors of cannabinoid agonist metabolism (e.g., without limitation, URB602, an inhibitor of monoacylglycerol lipase which catalyzes 2-arachidonoylglycerol hydrolysis) THC, nabilone, dronabinol, cannabidiol, 9-THC propyl analog, cannabidiol, cannabidiol propyl analog, cannabinol, cannabichromene, cannabichromene propyl analog, cannabigerol, cannabinoid terpenoids, cannabinoid flavonoids, endocannabinoids, anandamide, (R)- methanandamide,and 2-arachidonoylglycerol, THC-like ABC tricyclic cannabinoid analogues, exemplified by HU210 and desacetyllevonantradol; synthetic AC bicyclic and ACD tricyclic cannabinoid analogues, exemplified • by CP55940, and CP55244 and aminoalkylindole compounds, exemplified by WIN55212-2, and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof. (Little et al., Pharmacol. Biochem. Behav, 1989;Howlett et al, Neuropharmacology, 1990;Johnson et al, In: Cannabinoids as Therapeutic Agents (Mechoulam, R., ed.), CRC Press, 1986; Howlett et al., MoI Pharmacol, 1988; D'Ambra et al., J Med Chem, 1992; Pacheco et al., J Pharmacol Exp Ther, 1991; Compton et al, J Pharmacol Exp Ther, 1992; Howlett et al, Pharmacol Rev, 2002; Fowler. Fundam Clin Pharmacol. 2006;20: 549-62; Karanian and Bahr, Curr MoI Med 2006;6:677- 84; Singh and Budhiraja, Methods Find Exp Clin Pharmacol 2006;28: 177-83;
Mackie and Stella, AAPS J 2006;8:E298-306; Pavlopoulos, Curr Pharm Des 2006;12(14): 1751-69.).
[00183] In certain embodiments, the cannabinoid agonist useful for the present invention may be selected from the group consisting of dexanabinol (HU211), BAY 38-7271 , Naphthalen- 1 -yl-(4-pentyloxynaphthalen- 1 -yl)methanone, THC (delta-9-tetrahydrocannabinol), nabilone, dronabinol, cannabidiol, cannabinol, cannabichromene, cannabigerol, cannabigerol, anandamide, (R)- methanandamide, 2-arachidonoylglycerol, HU210, desacetyllevonantradol, CP55940, CP55244, URB602, or WIN55212-2 and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
[00184] In certain embodiments, the cannabinoid agonist useful for the present invention may be selected from the group consisting of 9-THC propyl analog, endocannabinoids, cannabinoid terpenoids, cannabinoid flavonoids, inhibitors of cannabinoid agonist metabolism, inhibitors of monoacylglycerol lipase, cannabidiol propyl analogues, cannabichromene propyl analogues, THC-like ABC tricyclic cannabinoid analogues, synthetic AC bicyclic cannabinoid analogues, synthetic ACD tricyclic cannabinoid analogues, aminoalkylindole compounds or analogs of 2-Arylimino-5,6-dihydro-4H-l,3-thiazines and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
[00185] In certain embodiments, the amount of the cannabinoid agonist used by the subject may be from about 10 ng to about 2000 mg, even up to about 2000 mg. More preferably, the amount of the cannabinoid agonist is from about 10 ng to about 1500 mg, even more preferably from about 0.1 mg to about 1000 mg, and most preferably, from about 0.1 mg to about 700 mg.
[00186] In some preferred embodiments, the cannabinoid agonist may be selected from compounds disclosed in U.S. Patent No. 7,217,732, 7,214,716,
7,169,942, 7,109,216, 7,091,216, 7,057,051 , 6,995, 184, 6,972,295, 6,943,266, 6,903, 137, 6,864,291, 6,864,285, 6,525,087, 6,524,805, 6.509.367. 6,284,788, 5,948,777, 5,939,429, and 5,605,906, and in U.S. Patent Application No. 20070167514, 20070123505, 20070105914, 20070099947, 20070088058, 20070088025, 20070087390, 20070060638, 20070032517, 20070027144, 20060293299, 20060241 165, 20060172019, 20060106071, 20060089356, 20060079557, 20060074086, 20050272763, 20050267161, 20050245554, 20050239828, 20050239133, 20050234061, 20050203112, 20050182103, 200501651 18, 20050154202, 20050137173, 20050101542, 20050096379, 20050065189, 20050054679, 20050026986, 20050009902, 20040266861, 20040266841 , 20040248956, 20040242593, 20040235854, 20040229928, 20040229850, 20040171613, 20040106614, 20040058820, 20040044051 , 20040034090, 20040018151, 20030175822, 20030138508, 200301 14495, 20020173528, 20020128302, 20020077322, and 20010034344, and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof. All of the above patents and patent applications are hereby incorporated by reference in their entirety.
[00187] For purposes of the present invention, the term "cannabinoid agonist" shall include combinations of more than one cannabinoid agonist, and also include the unsalifϊed agonist, mixed agonist-antagonists, partial agonists, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
[00188] Notwithstanding the above definitions of "cannabinoid agonist", for the purposes of the present invention, 1) drugs that enhance the effect of cannabinoid agonists by inhibiting their metabolism or reuptake (for example, anandamide amidase inhibitors) are considered to be cannabinoid agonists; 2) drugs that induce anandamide amidase inhibitor metabolism or induce CBj, CB2 and non-CB|/non-CB2 cannabinoid agonist metabolism or enhance reuptake will be considered cannabinoid antagonists; 3) inverse cannabinoid agonists will be considered cannabinoid antagonists.
[00189] Cannabinoid antagonists are known or readily determined by individuals who practice the art. Preferably, the cannabinoid antagonist useful for the present invention may be selected from the group consisting of SR 141716A [Rimonabant or N-piperidino-5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-4-methyl-3-pyrazole-carboxamide]), AM251, AM 281 ([N- moφholin-4-yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3- carboxamide), AM630, SR 144528 ([N-[(lS)-endo- 1,3,3- trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3-methyl-phenyl)- 1 -(4- methylbenzyl)pyrazole-3 -carboxamide]), 5-(4-chlorophenyl)- 1 -(2,4- dichlorophenyl)-3 -hexyl- 1 h- 1 ,2,4-triazole, 8-chloro- 1 -(2',4'-dichlorophenyl)- N-piperidin- 1 -yl- 1 ,4,5,6-tetrahydrobenzo[6,7]cyclohepta[ 1 ,2-c]pyrazole-3- carboxamide 4a, pyrazole class cannabinoid antagonists (e.g., SR141716 and SRl 44528), aminoalkylindole class cannabinoid antagonists (e.g., AM630), imidazolinedione class cannabinoid antagonists and triazole class cannabinoid antagonists, pyridone derivative class cannabinoid antagonists, quinolone derivative class cannabinoid antagonists, tricyclic derivatives of 1- benzylpyrazole-3-carboxylic acid, HU-308, HU-210, cannabidiol, tricyclic pyrazoles, analogs of 8-chloro- 1 -(2',4'-dichlorophenyl)-N-piperidin- 1 -yl- l,4,5,6-tetrahydrobenzo[6,7]cyclohepta[l,2-c]pyrazole-3-carboxamide, 5-(4- chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
[00190] For purposes of the present invention, the term "cannabinoid antagonist" shall include combinations of more than one cannabinoid agonist, and also include the unsalified agonist, mixed agonist-antagonists, partial agonists, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
[00191] In certain embodiments, the amount of the cannabinoid antagonist in the composition may be from about 10 ng to about 1000 mg, even up to about 2000 mg. More preferably, the amount of the cannabinoid antagonist is from
about 10 ng to about 1200 mg, even more preferably from about 0.1 mg to about 1000 mg, and most preferably, from about 0.1 mg to about 700 mg.
[00192] In some preferred embodiments, the cannabinoid antagonist may be selected from compounds disclosed in U.S. Patent No. 7,247,628, 7,176,210, 7,153,997, 7, 151,097, 7,132,414, 7,1 19, 108,6,930, 122, 6,642,258, 7,094,794, 6,916,838, 6,894,050, 6,875,782, 6,825, 198, 6,734,176, 6,673,802, 6,630,507, 6,555,578, 6,509,367, 6,344,481, 6,344,474, 6,194,454, 6,100,259, 5,989,583, 5,939,429, 5,747,524, 5,596,106, and 4,205,952, and in U.S. Patent Application No. 20070167514, 20070123505, 20070105914, 20070099947, 20070088058, 20070088056, 20070087390, 20070072907, 20070060638, 20070032517, 20070004772, 20060293299, 20060287341, 20060270655, 20060172019, 20060167049, 20060128673, 20060106071 , 20060100208, 20060100205, 20060089356, 20060079556, 20050272763, 20050267161, 20050267155, 20050250769, 20050245554, 20050239828, 20050239133, 20050234061 , 200502031 12, 20050187253, 20050182103, 20050154202, 20050101542, 20050096379, 20050065189, 20050054679, 20050026986, 20040266841 , 20040248956, 20040248944, 20040242593, 20040235854, 20040152736, 20040122074, 20040106800, 20040106614, 20040058820, 20040039024, 20040006105, 20030175822, 200301 14495, 20030087933, 20020188007, 20020128302, and 20020026050, and their or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof. All of the above patents and patent applications are hereby incorporated by reference in their entirety.
[00193] For purposes of the present invention, the term "cannabinoid antagonist" shall include combinations of more than one cannabinoid antagonist, and also include the unsalified antagonist, mixed agonist- antagonists, partial agonists, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
[00194] It is known that when coadministered with cannabinoid agonists, cannabinoid antagonists block the euphoric, pleasurable, reinforcing or toxic
effects of the cannabinoid agonist. Furthermore, it is known that cannabinoid antagonist administration in the setting of dependence or pharmacologic tolerance to cannabinoid agonists results in aversive effects, which may include signs and symptoms of cannabinoid agonist withdrawal. [00195] In certain embodiments, the effect of the cannabinoid agonist is at least partially blocked by the cannabinoid antagonist. In certain other embodiments, the effect of the cannabinoid agonist is substantially blocked by the cannabinoid antagonist. In certain embodiments, the cannabinoid antagonists precipitates signs or symptoms cannabinoid agonist withdrawal or abstinence in individuals who have developed tolerance to the cannabinoid agonist. In certain embodiments, the cannabinoid antagonist precipitates aversive effects of cannabinoid agonist withdrawal which discourage future abuse of the dosage form.
Alcohol Deterrents
[00196] A frequently co-abused substance in recreational drug users and drug addicts, including heroin users is alcohol.
[00197] One novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target other co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
[00198] The observation of polydrug abuse can be exploited to deter opioid agonist abuse indirectly by antagonizing the pleasurable and mood altering effects of alcohol which are not part of the dosage form of the invention but may nevertheless co-abused by opioid agonist abusers.
[00199] The term "alcohol deterrent" or "alcohol deterrence" means a molecule that causes a specific adverse physiologic, pathophysiologic or pharmacologic adverse effect to a human in the presence of alcohol or reduces the desire for alcohol.
[00200] All kinds of aversive effects of the alcohol deterrent in the presence of alcohol are anticipated, including a reduced desire for alcohol, nullification of mood altering and pleasurable effects, cutaneous flushing, vasodilation, throbbing, headache, respiratory distress, nausea, vomiting, retching, sweating,
thirst, chest pain, hypotension, orthostatic hypotension, dizziness, syncope, anxiety, uneasiness, weakness, vertigo, blurred vision, confusion, facial flush, pallor and/or shock.
[00201] In certain embodiments, when the dosage form is tampered, the alcohol deterrent precipitates aversive effects of alcohol which discourages future abuse of the dosage form containing the cannabinoid agonist. For the purposes of the present invention, the alcohol deterrent may also block the mood altering, euphoric, pleasurable, reinforcing, rewarding or toxic effects of the cannabinoid agonist upon tampering of the dosage form.
[00202] Alcohol deterrents are known or readily determined by individuals who practice the art. Alcohol deterrents include disulfiram, calcium carbimide, acmaprosate, diethylthiomethylcarbamate, inhibitors of aldehyde dehydrogenase, metabolites of disulfiram, metronidazole, chlorpropamide, topiramate, opioid receptor antagonists including naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan nalorphine, nalmefene, nadide, levallorphan, N-methylnaltrexone, N- allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N- allyllevallorphan.
[00203] In embodiments where the oral opioid agonist is in releasable form and the formulation further includes as an aversive agent an alcohol deterrent(s), said dosage forms having reduced potential for abuse in polydrug abusers. The invention does not provide abuse deterrence from the sequestered alcohol deterrent of the dosage form on the releasable opioid agonist of the dosage form under conditions of normal use (when administered whole or intact). However, under conditions of abuse or tampering (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered alcohol deterrent on: (i) co-used or co-abused alcohol, particularly in the setting of polydrug abuse; (ii) nullification of the mood altering effects of the opioid agonist in the presence of alcohol.
[00204] In certain embodiments, the amount of the alcohol deterrent in the claimed composition may be from about 10 ng to about 1000 mg, even up to about 2000 mg. More preferably, the amount of the alcohol deterrent is from about 10 ng to about 1200 mg, even more preferably from about 0.1 mg to about 1000 mg, and most preferably, from about 0.1 mg to about 700 mg.
Sequestered Cannabinoid Antagonists with Co-abused Cannabinoid Agonists
[00205] A frequently co-abused substance in recreational drug users and drug addicts, including heroin users is cannabis.
[00206] One novel method of deterring or minimizing opioid agonist misuse, abuse and tampering is to target other co-abused drugs that are not part of the abuse deterrent dosage form but which are frequently found in the systemic circulation of drug abusers.
[00207] The observation of polydrug abuse can be exploited to deter opioid agonist abuse indirectly by antagonizing the pleasurable and mood altering effects of pharmaceutical and non-pharmaceutical grade cannabinoids which are not part of the dosage form of the invention but may nevertheless co- abused by opioid agonist abusers.
[00208] Cannabinoid agonist abuse in the setting of polydrug abuse from the tampering of immediate release and particularly extended or sustained release formulations can be minimized by combining the releasable opioid agonist with a non-releasable or substantially non-releasable (i.e., sequestered) cannabinoid antagonist in the same dosage form, such that upon tampering, the cannabinoid antagonist becomes releasable, thereby: (i) reducing or eliminating the psychic effects of any co-abused cannabinoid agonist desired drug addicts and recreational drug users; and (ii) reducing or eliminating the toxic effects of the combination of the opioid agonist and the co-abused cannabinoid agonist in drug addicts and recreational drug users.
Opioid Agonists/Antagonists
[00209] The term "opioid receptor" includes mu (μ), delta (δ), kappa (K) and/or nociceptin/orphanin FQ (N/OFQ) peptide (NOP) receptors, their subtypes and splice variants such as μi, μ2, δi, δ2, Ki, K2 and K3, etc, regardless of whether
they also bind to or influence other receptor systems (e.g., norepinephrine reuptake inhibition, serotonin reuptake inhibition, NMDA receptor antagonism).
[00210] For the purposes of this invention, the term "opioid" is interchangeable with the term "opioid agonist", except when there is a specific reference to an opioid antagonist.
[00211] Opioid agonists include alfentanil, allylprodine, alphaprodine, anileridine, apomorphine, apocodeine, benzylmorphine, bezitramide, brifentanil, buprenorphine, butorphanol, carfentanil, clonitazene, codeine, cycloφhen, cyprenorphine, desomoφhine, dextromoramide, dezocine, diampromide, dihydrocodeine, dihydromoφhine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxyaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmoφhine, etonitazene, fentanyl, heroin, hydrocodone, hydroxymethylmoφhinan, hydromoφhone, hydroxypethidine, isomethadone, ketobemidone, levallor- phan, levoφhanol, levophenacylmoφhan, lofentanil, meperidine, meptazinol, metazocine, methadone, methylmoφhine, metopon, mirfentanil, moφhine, moφhine-6-glucuronide, myrophine, nalbuphine, narceine, nicomoφhine, norlevoφhanol, normethadone, nociceptin/oφhanin FQ (N/OFQ), normoφhine, noφipanone, ohmefentanyl, opium, oxycodone, oxymoφhone, papaveretum, pentazocine, phenadoxone, phenomoφhan, phenazocine, phenoperidine, pholcodine, piminodine, piritramide, propheptazine, promedol, profadol, properidine, propiram, propoxyphene, remifentanil, sufentanil, tapentadol, tramadol, trefentanil, tilidine, or any opioid having agonist activity at an opioid receptor belonging to the phenanthrene, moφhinan, benzomoφhan, methadone, phenylpiperidine, propionanilide 4- anilidopiperidine, 4-aryl piperidines, and 4-Heteroarylpiperidines class, any opioid having agonist activity at an opioid receptor having the same pentacyclic nucleus as nalmefene, naltrexone, buprenoφhine, levoφhanol, meptazinol, pentazocine and dezocine, any drug having agonist activity at an opioid receptor which is a fentanyl analog, or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs,
hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof. Opioid agonists also include drugs that bind to opioid receptors to exert agonist activity and are listed in the United States Controlled Substances Act of 1970, as amended, and regulations thereof, and drugs listed in the United States Psychotropic Substances Act of 1978, as amended, and regulations thereof.
[00212] The term "opioid antagonist" or "opioid receptor antagonist" means an antagonist substance that binds to one or more opioid receptor to exert an antagonist effect.
[00213] Opioid antagonists are known or readily determined by individuals who practice the art. Preferably, the opioid antagonists useful for the present invention may be selected from the group consisting of naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan nalorphine, nalmefene, nadide, levallorphan, N-methylnaltrexone, N- allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N- allyllevallorphan.
[00214] In certain embodiments of the present invention, the ratio of the opioid agonist and the cannabinoid antagonist, present in a substantially non- releasable form, is about 1: 1000 to about 1000: 1 by weight or about 1: 1000 to about 1000: 1, or about 1: 100 or about 100: 1 by weight, preferably about 1 : 10 to about 10:1 by weight, and more preferably about 5: 1 to 1:5 by weight. The weight ratio of the opioid agonist to cannabinoid antagonist, as used in this application, refers to the weight of the active ingredients.
[00215] The present invention provides an oral dosage form of opioid agonist useful for decreasing the potential for abuse of the opioid agonist contained therein. The present invention includes an oral dosage form comprising an orally therapeutically effective amount of an opioid agonist in combination with a cannabinoid antagonist. The cannabinoid antagonist is present in a substantially non-releasable form'.
[00216] In some embodiments, the present invention relates to oral opioid agonist in releasable form and cannabinoid antagonist in substantially non- releasable form (i.e., sequestered) when administered intact, said dosage forms
having reduced potential for abuse in opioid abusers and polydrug abusers. In some embodiments, the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered cannabinoid antagonist on co-abused cannabinoid agonists in the setting of polydrug abuse under conditions where the dosage form of the invention is abuse or tampered (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), said cannabinoid agonist not part of the dosage form and said dosage form having no significant direct effect on the opioid agonist of the dosage form when the dosage form is used as directed or, in some embodiments, even when subject to abuse, and on any co-used cannbinoid agonist when the dosage form of the invention is not abused or tampered.
[00217] In some embodiments, the present invention relates to oral opioid agonist in releasable form and an alcohol deterrent in substantially non- releasable form (i.e., sequestered) when administered intact, said dosage forms • having reduced potential for abuse in alcohol abusers and polydrug abusers. In some embodiments, the invention achieves its abuse deterrence by a novel method, namely by the effects of the sequestered alcohol deterrent on used or co-abused alcohol in the setting of polydrug abuse under conditions where the dosage form of the invention is abused or tampered (e.g., crushing or solvent extraction, followed by ingestion orally, inhalationally, intranasally or parenterally), said alcohol not part of the dosage form and said dosage form having no significant direct effect on the opioid agonist of the dosage form when the dosage form is used as directed or, in some embodiments, even when subject to abuse in the absence of alcohol, and on any co-used alcohol when the dosage form of the invention is not abused or tampered.
[00218] In certain embodiments, the cannabinoid antagonist present in a substantially non-releasable form does not substantially block the therapeutic effects of the opioid agonist when the dosage form is orally administered intact (and does not pose a risk of precipitation of withdrawal in cannabinoid tolerant or dependent patients), but wherein the effect of the opioid agonist is at least partially blocked by the cannbinoid antagonist when said dosage form
is tampered with, e.g., chewed, crushed or dissolved in a solvent, and administered orally, intranasally, by inhalation, parenterally or sublingually.
Sequestering the Aversive Agent
[00219] In certain preferred embodiments of the present invention, the substantially non-releasable form of the aversive agent comprises aversive agent particles in a coating that substantially prevents the release of the aversive agent. In preferred embodiments, the coating comprising one or more of pharmaceutically acceptable hydrophobic material. The coating is preferably impermeable to the cannabinoid antagonist contained therein and is insoluble in the gastrointestinal system, thus substantially preventing the release of the cannabinoid antagonist when the dosage form is administered orally as intended.
[00220] Although the preferred embodiments of the invention comprise a cannabinoid antagonist or alcohol deterrent in a form that completely prevents the release of the aversive agent, the invention also includes an aversive agent in a substantially non-releasable form. The term "substantially not released" N refers to the aversive agent that might be released in a small amount, as long as the amount released does not affect or does not significantly affect therapeutic efficacy of the agonist when the dosage form is orally administered to humans as intended.
[00221] In certain preferred embodiments of the invention, the substantially non-releasable form of the aversive agent is resistant to laxatives (e.g., mineral oil) used to manage constipation.
[00222] In certain embodiments, the substantially non-releasable form of the aversive agent is formulated with one or more of pharmaceutically acceptable hydrophobic material, such that the antagonist is not released or substantially not released during its transit through the gastrointestinal tract when administered orally as intended, without having been tampered with.
[00223] In certain embodiments of the present invention, the substantially non- releasable form of the aversive agent is vulnerable to mechanical, thermal and/or chemical tampering, e.g., tampering by means of crushing, shearing,
grinding, chewing, and/or dissolution in a solvent in combination with heating of the oral dosage form. When thus tampered with, the integrity of the substantially non-releasable form of the aversive agent will be compromised, and the aversive agent will be made available to be released. In certain embodiments, when the dosage form is chewed, crushed or dissolved and heated in a solvent, and administered orally, intranasally, inhalationally, parenterally or sublingually, the analgesic, euphoric, pleasurable, reinforcing or toxic effects of the cannabinoid is reduced or eliminated.
[00224] Accordingly, when the oral dosage form is not tampered with as to compromise the integrity of the coating, the aversive agent contained therein will not be substantially released during its first hour of transit through the gastrointestinal system, and thus would not be available for absorption. In certain preferred embodiments of the present invention, the hydrophobic material comprises a cellulose polymer or an acrylic polymer that is insoluble in the gastrointestinal fluids and impermeable to the aversive agent.
[00225] Accordingly, when the oral dosage form is not tampered with as to compromise the integrity of the coating, the aversive agent contained therein will not be substantially released during its transit through the gastrointestinal system, and thus would not be available for absorption.
[00226] The term "particles" of aversive agent, as used herein, refers to granules, spheroids, beads or pellets comprising the aversive agent. In certain preferred embodiments, the aversive agent particles are about 0.2 to about 2 mm in diameter, more preferably about 0.5 to about 2 mm in diameter.
[00227] In certain embodiments of the present invention, the oral dosage form further comprises an aversive agent (e.g., cannabinoid antagonist or alcohol deterrent) in a releasable form and is thus capable of being released from the oral dosage form when orally administered, the ratio of the opioid agonist to the releasable form of the aversive agent (e.g., cannabinoid antagonist) being such that the dosage form, when administered orally, is therapeutically effective as an opioid agonist. For example, when the aversive agent (e.g., cannabinoid antagonist) is coated with a coating that substantially prevents its release, and is then mixed with an opioid agonist and compressed into tablets,
certain amounts of the coating might be cracked, thus exposing the aversive agent (e.g., cannabinoid antagonist) to be released upon oral administration.
[00228] In certain embodiments of the present invention, the ratio of the opioid agonist to the aversive agent (e.g., a cannabinoid antagonist, or an alcohol deterrent) present in a substantially non-releasable form, is about 10000: 1 to about 1: 10000, or about 1000:1 to about 1: 1000 by weight, or preferably about 1 : 100 to 100: 1 by weight or about 1 : 10 to about 10: 1 by weight, and more preferably about 5: 1 to 1:5 by weight. The weight ratio of the opioid agonist to the aversive agent, as used in this application, refers to the weight of the active ingredients. Thus, for example, the weight of the aversive agent excludes the weight of the coating or matrix that renders the aversive agent substantially non-releasable, or other possible excipients associated with the aversive agent particles. Since the aversive agent is in a substantially non- releasable from, the amount of such aversive agent within the dosage form may be varied more widely, as the formulation does not depend on differential biotransformation or pharmacodynamics for proper functioning. For safety reasons, the amount of the aversive agent present in a substantially non- releasable form is selected as not to be permanently harmful to humans even if fully released by tampering with the dosage form.
[00229] In preferred embodiments, the oral dosage form of the present invention is directed to an oral dosage form comprising (i) a opioid agonist in releasable form and (ii) a sequestered aversive agent which is substantially not released when the dosage form is administered intact, such that the ratio of the mean Cmax of the aversive agent after single dose oral administration of the dosage form after tampering to the mean Cmax of aversive agent after single dose oral administration of an intact dosage form is at least 1.5: 1. In other embodiments of the invention, the mean Cmax ratio using the aforementioned test method is at least 3: 1, 6: 1, 10: 1, 20: 1, 30: 1, 40:1, 50: 1, 70: 1, 100: 1 or 500: 1.
[00230] Alternatively or additionally, preferably the ratio of the mean AUCo-t or AUCo-oo of the aversive agent after single dose oral administration of an immediate release reference product containing an equivalent amount of
aversive agent to the mean AUCo-1 or AUCo-∞ of aversive agent after single dose oral administration of an intact dosage form is at least 1.5:1. In other embodiments of the invention, the mean AUCo-t or AUCo-∞ ratio using the aforementioned test method is at least 3: 1, 6: 1, 10: 1, 20:1, 30: 1, 40: 1, 50: 1, 70: 1, 100: 1 or 500: 1.
[00231] Alternatively or additionally, the ratio of the mean Tmax of the aversive agent after single dose oral administration of the intact dosage form to the mean Tmax of aversive agent after single dose oral administration of a dosage form after tampering is at least 1.5: 1. In other embodiments of the invention, the mean Tmax ratio using the aforementioned test method is at least 3: 1, 6: 1, 10: 1 or 20: 1.
[00232] The oral dosage form containing a opioid agonist in combination with a substantially non-releasable form of an aversive agent includes, but are not limited to tablets or capsules.
[00233] The dosage forms of the present invention may include any desired pharmaceutical excipients known to those skilled in the art. Specific examples of pharmaceutically acceptable carriers and excipients that may be used to formulate oral dosage forms of the present invention are described in the Handbook of Pharmaceutical Excipients, APhA Publications; 5 edition (January 5, 2006), compounds found on the FDA EAFUS database (http://vm.cfsan.fda.gov/~dms/eafus.html); FDA Food Additives Status List (http://www.cfsan.fda.gov/~dms/opa-appa.html); FDAGRAS list and database; FDA Color Additive Status List
(http://www.cfsan.fda.gov/~dms/opa-appc.html); FDA Inactive Ingredients Database (http://www.accessdata.fda.gov/scripts/cder/iig/index.cfm); Rowe, Sheskey and Owen, Handbook of Pharmaceutical Excipients, APhA Publications; 5th edition (2006); Remington: The Science and Practice of Pharmacy, 21st ed, Lippincott Williams & Wilkins (2005); United States Pharmacopeia-National Formulary (USP-NF), (USP 30 - NF 25, 2007), the International Programme on Chemical Safety (http://www.inchem.org/) and Health Canada's List of Acceptable Non-medicinal Ingredients
(http://www.hc-sc.gc.ca/dhp-mps/prodnatur/legislation/docs/nmi- imn_listl_e.html), all hereby incoφorated by reference in their entirety.
[00234] The oral dosage forms may further provide an immediate release of the opioid agonist. In certain embodiments, the oral dosage forms of the present invention provide a sustained release of the opioid agonist contained therein. Oral dosage forms providing sustained release of the opioid agonist may be prepared in accordance with formulations/methods of manufacture known to those skilled in the art of pharmaceutical formulation, e.g., via the incorporation of a sustained release carrier into a matrix containing the substantially non-releasable form of an aversive agent; or via a sustained release coating of a matrix containing the opioid agonist and the substantially non-releasable form of the aversive agent.
[00235] The benefits of the abuse-resistant dosage form are especially great in connection with oral dosage forms of potent opioid agonists, which can provide valuable therapeutic benefits but are prone to being abused. This is particularly true for sustained release opioid agonist products which have a large dose of a desirable opioid agonist intended to be released over a period of time in each dosage unit. Drug abusers take such sustained-release product and crush, grind, extract or otherwise damage the product so that the full contents of the dosage form become available for immediate absorption. Since such tampering of the dosage form of the invention results in the cannabinoid antagonist also becoming available for absorption, the present invention provides a-means for deterring such abuse. In addition, the present invention addresses the risk of overdose to ordinary patients from "dumping" effect of the full dose of the opioid agonist if the product is accidentally chewed or crushed.
[00236] The invention may provide for a safer product (e.g., lower risk of opioid agonist toxicity or polydrug toxicity), if the product is misused, as well as one with less risk of abuse.
[00237] In certain embodiments, a combination of two opioid agonists is included in the formulation with the aversive agent. In further embodiments, one or more opioid agonist and an aversive agent is included and a further
non-opioid agonist drug is also included for the treatment of the same medical condition as the opioid agonist or for the treatment of a different medical condition. In certain embodiments, a combination of two or more aversive agents for interfering with the same or a different type of abuse (e.g., two cannabinoid antagonists; a cannabinoid antagonist and an alcohol deterrent are included in the formulation with the opioid agonist(s).
[00238] In yet other embodiments, the dosage form is co-administered with a non-opioid agonist drug for the treatment of the same medical condition as the opioid agonist or for the treatment of a different medical condition. All modes of co-administration are contemplated, including via oral, subcutaneous, direct intravenous, slow intravenous infusion, continuous intravenous infusion, intravenous or epidural patient controlled analgesia (PCA and PCEA), intramuscular, intrathecal, epidural, intracisternal, intramuscular, intraperitoneal, transdermal, topical, transmucosal, buccal, sublingual, transmucosal, inhalation, intranasal, epidural, intra-atricular, intranasal, rectal or ocular routes.
[00239] All oral pharmaceutical dosage forms of the invention are contemplated, including oral suspensions, tablets, capsules, lozenges, effervescent tablets, transmucosal films, buccal products, oral mucoretentive products and the like, administered as immediate release, sustained release, delayed release, modified release, controlled release, extended release and the like.
[00240] In certain embodiments, the dosage form may include, in addition to the releasable opioid agonist and the substantially non-releasable cannabinoid antagonist, other abuse deterrent substances in releasable or substantially non- releasable form, including various aversive agents know to practitioners of the art.
[00241] Depending upon the particular route of administration, a variety of pharmaceutically-acceptable carriers, well known in the art may be used. Nonlimiting examples are sugars, starches, cellulose and its derivatives, malt, gelatin, talc, calcium sulfate, vegetable oils, synthetic oils, polyols,
alginic acid, phosphate buffered solutions, emulsifiers, isotonic saline, and pyrogen-free water.
Additional Active Drugs
[00242] Other pharmaceutically active ingredients (drugs) from various therapeutic classes may also be used in combination with the present invention, e.g., included in the dosage forms of the invention. They include, but are not limited to decongestants, analgesics, analgesic adjuvants, antidepressants, antipsychotics, anxiolytics, hypnotics, sedatives, anti-ADHD drugs, psychostimulants, drugs to treat urinary incontinence, antihistamines, expectorants, antitussives, diuretics, anti-inflammatory agents, antipyretics, antirheumatics, antioxidants, laxatives, local anesthetics, proton pump inhibitors, motility modifying agents, vasodilators, inotropes, beta blockers, beta adrenergic agonists, drugs to treat asthma and COPD, antiinfectives, antimigraine agents, antihypertensives, antianginal agents, gastric acid reducing agents, anti-ulcer agents, anticoagulants, lipid and cholesterol lowering drugs, anti-diabetic drugs, anti-epileptics, hormones, smooth muscle relaxants, skeletal muscle relaxants, bronchodilators, vitamins, trace minerals, amino acids, biological peptides and drugs to treat various infectious, immunologic disorders, cardiovascular, pulmonary, gastrointestinal, hepatic, biliary, nutritional, metabolic, endocrine, hematologic, oncologic, musculoskeletal, neurologic, psychiatric, genitourinary, gynecologic, obstetric, pediatric, otolaryngogologic, ophthalmic, dermatologic, dental, oral, and genetic disorders, diseases and maladies. The drug being used in combination therapy with the present invention can be administered by any route, including parenterally, orally, topically, transdermally, sublingually, and the like. In certain preferred embodiments of the present invention, the invention allows for the use of lower doses of the opioid agonist by virtue of the inclusion of an additional non-opioid agonist drug for the prevention or treatment of the same medical condition. By using lower amounts of either or both drugs, the side effects associated with treatment in humans are reduced.
[00243] The present invention is further directed to a method of decreasing the potential for abuse of an opioid agonist in an oral dosage form. The method comprises providing the opioid agonist in an oral dosage form as described herein.
[00244] The present invention is directed to an immediate and controlled release opioid agonists which are formulated in order to reduce and minimize misuse, abuse and diversion. In certain embodiments, these characteristics are conferred by the inclusion of a cannabinoid antagonist or alcohol deterrent, which is itself formulated in a unique controlled release matrix. The properties of this formulation are developed to liberate the aversive agent in conditions of misuse or tampering yet a negligible amount of aversive agent would be released (an amount which does not affect therapeutic effect of the opioid agonist experienced by the patient) under the prescribed conditions of use.
[00245] In certain embodiments of the invention, the release for the aversive agent (e.g., antagonist component) of the formulation is expressed in terms of a ratio of the release achieved after tampering, e.g., by crushing or chewing, relative to the amount released from the intact formulation. The ratio is therefore expressed as [Crushed]/[Whole], and it is desired that this ratio have a numerical range of at least 4: 1 or greater (crushed release in 1 hour/intact release in 1 hour).
[00246] The present invention provides an oral dosage form of opioid agonist useful for decreasing the potential for abuse of the opioid agonist contained therein. The present invention includes an oral dosage form comprising an orally therapeutically effective amount of an opioid agonist in combination with an aversive agent (e.g., cannabinoid antagonist). The cannabinoid antagonist is present in a substantially non-releasable form.
[00247] In certain preferred embodiments, the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form comprises aversive agen particles coated with a coating that substantially prevents its release. In preferred embodiments, such coating surrounds the aversive agent particles and is impermeable to the drug and is insoluble in the gastrointestinal system. When the dosage form of the present invention is orally administered to
humans, the aversive agent (e.g., cannabinoid antagonist) is not substantially released from the coating and is, therefore, not available for absorption into the body. Thus, the aversive agent (e.g., cannabinoid antagonist), although present in the dosage form, does not substantially block the therapeutic effectiveness of the opioid agonist. However, if the oral dosage form of the present invention is tampered with as to compromise the integrity of the coating, the cannabinoid antagonist contained therein would be made available to at least partially block the effect of the opioid agonist. This characteristic decreases the potential for abuse or diversion of the opioid agonist in the oral dosage form. For example, if one attempts to abuse the drug contained in the oral dosage form of the present invention by, e.g., chewing, crushing, grinding or dissolving it in a solvent with heat (e.g., greater than about 45. degree. C. to about 50. degree. C), the coating will be damaged and will no longer prevent the cannabinoid antagonist from being released. Upon administration, the aversive agent (e.g., cannabinoid antagonist) will be released and significantly block the euphoric, pleasurable, reinforcing or toxic effects of the opioid agonist and of any co-used or co-abused cannabinoid agonist.
[00248] In certain embodiments of the invention, the ratio of the opioid agonist to the coated aversive agent (e.g., cannabinoid antagonist) is such that when the oral dosage form is tampered with as to compromise the integrity of the coating that renders the aversive agent (e.g., cannabinoid antagonist) substantially non-releasable, the euphoric, pleasurable, reinforcing or toxic effects of the agonist would be negated by the cannabinoid antagonist when misused by a human subject orally, parenterally, intranasally, inhalationally or sublingually. In certain preferred embodiments of the invention, the euphoric, pleasurable, reinforcing or toxic effects of the opioid agonist would be negated by the aversive agent (e.g., cannabinoid antagonist) when misused parenterally or sublingually.
[00249] The present invention also includes an oral dosage form which comprises a releasable form of an averdive agent (e.g., cannabinoid antagonist), along with a opioid agonist and coated aversive agent (e.g., cannabinoid antagonist) particles, the ratio of the agonist to the non-coated
aversive agent (e.g., cannabinoid antagonist) being such, when administered orally as intended, the oral dosage form is therapeutically or analgesically effective.
[00250] In certain other embodiments of the present invention, the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form comprises an aversive agent (e.g., cannabinoid antagonist) dispersed in a matrix that renders the antagonist substantially non-releasable, wherein the matrix comprises one or more of a pharmaceutically acceptable hydrophobic material. The aversive agent (e.g., antagonist) is substantially not released from the matrix, thus is not made available to be absorbed during its transit through the gastrointestinal system.
[00251] In certain other embodiments of the present invention, the aversive agent (e.g., cannabinoid antagonist) in a matrix that renders the aversive agent (e.g., antagonist) substantially non-releasable comprises an aversive agent (e.g., cannabinoid antagonist) dispersed in a melt-extruded matrix, wherein the matrix comprises one or more of a pharmaceutically acceptable hydrophobic material.
[00252] The oral dosage form of the present invention may further include, in addition to a opioid agonist and aversive agent (e.g., antagonist), one or more drugs that may or may not act synergistically therewith. Thus, in certain embodiments, a combination of two opioid agonists may be included in the dosage form, in addition to the aversive agent (e.g., cannabinoid antagonist). For example, the dosage form may include two opioid agonists having different properties, such as half-life, solubility, potency, and a combination of any of the foregoing. In yet further embodiments, one or more opioid agonist is included and a further non- opioid agonist drug is also included, in addition to the aversive agent (e.g., cannabinoid antagonist). In a further embodiment, a non-opioid agonist drug is also included for the treatment of the same medical condition as the opioid agonist or for a different medical condition. In some embodiments, the opioid agonist is intended to prevent or treat acute or chronic pain. An included non- opioid agonist drug in such a dosage form may be used to provide additive, complementary, or synergistic therapeutic
effects, including NSAIDs, NO-NSAIDs, COX-2 selective inhibitors, acetaminophen, nitroparacetamol, nitric oxide donors, tramadol, beta adrenergic agonists, alpha-2 agonists, selective prostanoid receptor antagonists, cannabinoid agonists, opioid receptor agonists, NO-opioid receptor agonists, local anesthetics, purinergic P2 receptor antagonists, NMDA receptor antagonists, gabapentin, pregabalin, gabapentinoids, ligands of alpha(2)delta subunits of voltage-gated calcium channels, neuronal nicotinic receptor agonists, calcium channel antagonists, sodium channel blockers, superoxide dismutase mimetics, p38 MAP kinase inhibitors, TRPVl agonists, dextromethorphan, dextrorphan, ketamine, glycine receptor antagonists, antidepressants, corticosteroids, and antiepileptics, and any other drugs that can be shown by a person proficient in the art to prevent or treat pain.
Additional Aversive Agent - Abuse Intervention Agents
[00253] In certain preferred embodiments of the invention, the dosage form optionally comprises, in addition to the foregoing opioid agonist and the aversive agent(s) (selected from cannabinoid antagonists and alcohol deterrents, and combinations of the same), one of more additional agents that are referred to herein as an abuse intervention agent(s), in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form. The abuse intervention agent(s) may comprise, for example, laxatives, cutaneous vasodilators, headache producing agents, emetics, emetogenic compound, nausea producing compounds, bittering agents, drugs that cause burning on irritation when in contact with tissue or mucous membranes (e.g., naso-mucosal irritants, oro-mucosal irritants, respiratory irritants), tissue irritants, gastrointestinal irritants, drugs that precipitate withdrawal effects, tissue dyes, lakes and colorants, beverage dyes, lakes and colorants, non-tissue staining beverage dyes, lakes and colorants (i.e, that do not stain or discolor the skin upon ingestion), fecal discolorants, urine discolorants, malodorous agents, opioid antagonists, benzodiazepine antagonists (e.g., flumazenil), and the like.
[00254] In certain particularly preferred embodiments of the invention, the abuse intervention agents are further selected from the group comprising (i) laxatives; (ii) cutaneous vasodilators; (iii) headache producing agents; (iv) emetics, emetogenic and nausea producing compounds; (iv) bittering agents (v) mucosal, naso-mucosal, oro-mucosal, respiratory, tissue and gastrointestinal irritants; (vi) tissue staining, non-tissue staining and beverage staining dyes, lakes and colorants; (vii) fecal and urine discolorants; (viii) malodorous agents; (ix) opioid antagonists; and (x) benzodiazepine antagonists (e.g., flumazenil), and mixtures thereof.
[00255] In certain particularly preferred embodiments of the invention, the abuse intervention agent comprises a non-toxic dye to deter surreptitious attempts at intoxication of another subject (e.g., in an alcoholic or nonalcoholic beverage).
[00256] In certain particularly preferred embodiments of the invention, the dosage form comprises an abuse intervention agent which comprises a nontoxic bittering agent to deter surreptitious attempts at intoxication of another subject (e.g., in an alcoholic or non-alcoholic beverage).
[00257] In certain particularly preferred embodiments of the invention, the dosage form comprises an abuse intervention agent which comprises a nontoxic bittering agent to deter oral or nasal ingestion of the dosage form.
[00258] In certain particularly preferred embodiments of the invention, the dosage form comprises an abuse intervention agent which comprises a nontoxic nasal irritant to deter oral or nasal ingestion of the dosage form.
[00259] In some embodiments, the abuse intervention agent(s) may be in the dosage form in an amount that does not produce an aversive effect or aversion in any, many or substantially all patients when taken in accordance with the prescribing information or the manufacturer's instructions (for example, in small quantities), but which produce an aversive effect when taken in excess (e.g., higher dose or more frequently).
[00260] In some embodiments, the abuse intervention agent is one or more bittering agents selected from the group comprising T2R or TAS2R receptor agonists, phenylthiourea (phenylthiocarbamide), natural, artificial and
synthetic flavor oils, flavoring aromatics, flavoring oils, oleoresins, spearmint oil, peppermint oil, eucalyptus oil, oil of nutmeg, allspice, mace, oil of bitter almonds, menthol, citrus oils including lemon, orange, lime, grapefruit, and fruit essences, sucrose derivatives, sucrose octaacetate, chlorosucrose derivatives, quinine, denatonium, denatonium saccharide and denatonium benzoate.
[00261] In some embodiments, the abuse intervention agent is one or more naso-mucosal, oro-mucosal, respiratory or tissue irritants selected from the group comprising transient receptor potential vanilloid 1 agonists, resiniferanoids, capsaicinoids, phorboid vanilloids, terpenoid 1,4-unsaturated dialdehydes, capsaicin, capsaicin analogs, resiniferatoxin, olvanil, pipeline, zingerone, anandamide, 12- and 15-(S)-hydroperoxy-eicosatetraenoic acids, 5 -•and 15-(S)-hydroxyeicosatetraenoic acids, phorbol 12-phenylacetate 13- acetate 20-homovanillate, 2 phorbol 12,13-didecanoate 20-homovanillate, leukotriene B(4), tinyatoxin, heptanoylisobutylamide, N-(3-acyloxy-2- benzylpropyl)-N'-dihydroxytetrahydrobenzazepine, tetrahydroisoquinoline thiourea analogs, heptanoyl guaiacylamide, isobutylamides, guaiacylamides, dihydrocapsaicin, homovanillyl octylester, nonanoyl vanillylamide, formic acid, acetic acid, propionic acidy, butyric acid, valeric acid, caproic acid, caprillic acid, capric acid, oxalic acid, malonic acid, succicnic acid, glu~taric acid, adipic acid, maleic acid, fumaric acid, citric acid, sodium lauryl sulfate, poloxamer, sorbitan monoesters, glyceryl monooleates, niacin, mustard, allyl isothiocyaanate and p-hydroxybenzyl isothiocyanate and acetylsalicylic acid.
[00262] In some embodiments, the abuse intervention agent is one or more emetogenic or nausea producing agents selected from the group comprising zinc and pharmaceutically acceptable salts thereof, dopamine agonists, apomorphine, ipecac, ipecacuanha, emetine, methylcephaeline, cephaeline, psychotrine, O-methylpsychotrine, ammonium chloride, potassium chloride, magnesium sulfate, ferrous gluconate, ferrous sulfate, aloin, algarot or antimonious oxychloride, antimony trichloride, folate, folic acid, niacin and nicotinamide.
[00263] In some embodiments, the abuse intervention agent is one or more cutaneous vasodilators selected from the group comprising niacin, nicotinuric acid, beta-hydroxybutyrate and nicotinic receptor agonists, including agonists at nicotinic receptor HM74A and nicotinic receptor GPR 109 A.
[00264] In some embodiments, the abuse intervention agent is one or more tissue dyes, lakes or colorants, or beverage dyes, lakes or colorants, or a beverage dye, lake and colorant that does not stain or discolor the skin upon ingestion, or a fecal discolorant or a urine discolorant selected from the group comprising Curcumin, Riboflavin, Tartrazine, Quinoline yellow, Sunset yellow FCF, Carmine, Carmoisine, Amaranth, Ponceau 4R, Erythrosine, Allura red AC, Patent blue V, Indigo carmine, Brilliant blue FCF, Chlorophylls, Copper complexes of chlorophylls and chlorophyllins, Green S, Caramel, Brilliant black BN, Vegetable carbon, Carotenoids, Alpha-, beta-, gamma-carotene, Capsanthin, Capsorubin, Lycopene, Beta-apo-8' carotenal, Ethyl ester of beta-apo-8' carotenoic acid, Xanthophylls, Lutein, Canthaxanthin, Beetroot red, Anthocyanins, Cyanidin, Delphidin, Malvidin, Pelargonidin, Peonidin, Petunidin, Calcium carbonate, Titanium dioxide, Iron oxides and hydroxides, Aluminum, Brilliant blue FCF, Indigotine, Alphazurine FG, Indanthrene blue, Fast green FCF, Alizarin cyanine green F, Quinizarine green SS, Pyranine concentrated, Orange II, Dibromofluorescein, Diiodofluorescein, Erythrosine yellowish Na, Erythrosine, Ponceau SX, Lithol rubin B, Lithol rubin B Ca, Toney red, Tetrabromofluorescein, Eosine, Tetrachlorotetrabromofluorescein, Phloxine B, Helindone pink CN, Brilliant lake red R, Acid fuchsine, Lake bordeaux B, Flaming red, Alba red, Allura red AC, Allura Red AC, Alizurol purple SS, Tartrazine, Sunset yellow, FCF, Fluorescein, Naphthol yellow S, Uranine, Quinoline yellow WS, Quinoline yellow SS, Brilliant blue FCF, Indigotine, Alphazurine FG, Alizurol purple SS, Sunset yellow FCF, Alumina, Aluminum powder, Annatto extract, Beta- carotene, Bismuth oxychloride, Bronze powder, Calcium carbonate, Canthaxanthin, Caramel, Chromium-cobalt-aluminum oxide, Chromium hydroxide green, Chromium oxide green, Cochineal extract, carmine, Copper powder, Dihydroxyacetone, Ferric ammonium citrate, Ferric ammonium
ferrocyanide, Ferric ferrocyanide, Guanine, Iron oxides synthetic, Logwood extract, Mica, Potassium sodium copper chlorophyllin, Pyrogallol, Pyrophyllite, Talc, Titanium dioxide, Zinc oxide, FD&C blue #1, FD&C blue #2, D&C blue #4, D&C blue #9, FD&C green #3, D&C green #5, D&C green #6, D&C green #8, D&C orange #4, D&C orange #5, D&C orange #10, D&C orange #11, FD&C red #3, FD&C red #4, D&C red #6, D&C red #7, D&C red #17, D&C red #21, D&C red #22, D&C red #27, D&C red #28, D&C red #30, D&C red #31, D&C red #33, D&C red #34, D&C red #36, D&C red #39, FD&C red #40, FD&C red #40 lake, D&C violet #2, FD&C yellow #5, FD&C yellow #6, D&C yellow #7, Ext. D&C yellow #7, D&C yellow #8, D&C yellow #10, D&C yellow #11, FD&C lakes, D&C lakes, Ext. D&C lakes, FD&C blue #1 lake, FD&C blue #2 lake, D&C blue #4 lake, FD&C green #3 lake, D&C green #5 lake, D&C green #6 lake, D&C orange #4 lake, D&C orange #5 lake, D&C orange #10 lake, D&C orange #11 lake, FD&C red #4 lake, D&C red #6 lake, D&C red #7 lake, D&C red #17 lake, D&C red #21 lake, D&C red #22 lake, D&C red #27 lake, D&C red #28 lake, D&C red #30 lake, D&C red #31 lake, D&C red #33 lake, D&C red #34 lake, D&C red #36 lake, D&C violet #2 lake, FD&C yellow #5 lake, FD&C yellow #6 lake, D&C yellow #7 lake, Ext. D&C yellow #7 lake, D&C yellow #8 lake, D&C yellow #10 lake, Turmeric, Lacto flavin, Cochineal, carminic acid, Indigotine, Magnesium chlorophyll, Brilliant green BS, Black PN, Carbo medicinalis vegetabilis, Paprika oleoresin, Paprika oleoresin, Betanin, Beta-carotene, indigo carmine, iron oxides, sunset yellow FCF, titanium dioxide, ElOO, ElOl, E102, E104, EI lO, E120, E122, E123, E124, E127, E129, E131, E132, E133, E140, E141, E142, E150, E151, E153, E160, E161, E162, E163, E170, E171, E172, E173 and phenazopyridine. In some embodiments, the abuse intervention agent is one or more laxatives selected from the group comprising Bis(p-hydroxyphenyl)pyridyl-2- methane, bisacodyl, bisoxatin, anthraquinone, anthraquinone analogs and derivatives (e.g., buckthorn, casanthranol, cascara, hydroxyanthracene, glucofrangulin ), dantron, danthron, docusate (e.g., docusate sodium, docusate calcium, docusate potassium), gastrointestinal chloride channel activators
(e.g., chloride channel subtype 2 activators), lubiprostone, magenesium salts (e.g., magnesium citrate, magnesium hydroxide, magnesium oxide), mannitol, oxyphenisatine, polyethylene glycol, polyethylene oxide) [PEO- 1500], sodium phosphate, phenolphthalein, senna, senna constituents and derivatives (e.g., sennoside A, sennoside B) and sodium picosulfate.
[00266] In some embodiments, the abuse intervention agent is one or more opioid antagonists selected from the group comprising naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan nalorphine, nalmefene, nadide, levallorphan, N-methylnaltrexone, N- allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N- allyllevallorphan.
[00267] In some embodiments, the abuse intervention agent may be added to the formulation in an amount of less than about 80% by weight, preferably less than about 60% by weight, more preferably less than about 40% by weight of the dosage form, even more preferably less than about 20% by weight of the dosage form, and most preferably less than about 10 by weight of the dosage form (e.g., 0.000000000000001% to 1%, or 0.000000001% to 3%, or 0.0001% to 10%, or 0.001% to 5%, or 1% to 10%, or 0.001% to 2%, or 1% or 10%, or 2% to 7%) depending on the particular aversive agent used.
[00268] In some embodiments, the abuse intervention agent in the dosage form may be about 0.00000000001 mg to about 2000 mg, or about 0.0000001 mg to about 1500 mg, or about 0.000001 mg to about 1000 mg, or about 0.0001 mg to about 1000 mg, or about 0.001 mg to about 1000 mg, or about 0.01 mg to about 1000 mg, or about 0.1 mg to about 1500 mg, or 1 mg to about 800 mg, or about 1 mg to about 500 mg, or about 1 mg to about 300 mg, or about 1 mg to about 150 mg, or about 5 mg to about 400 mg, or about 5 mg to about 200 mg, or about 0.00000000001 mg to about 200 mg, or about 0.00000000001 mg to about 100 mg, or about 0.00000000001 mg to about 50 mg, or about 0.0000001 mg to about 200 mg, or about 0.0000001 mg to about 100 mg, or about 0.00001 mg to about 400 mg, or about 0.0001 mg to about 300 mg.
[00269] In some embodiments, the amount of the abuse intervention agent in the dosage form of the present invention can be a fixed ratio in relation to the
amount of opioid agonist in the dosage form. By appropriately selecting the quantity of the other aversive agent in the dosage form, aversive effects can be avoided under conditions of proper medical use (e.g., manufacturers prescribing directions). However, under some conditions of abuse, for example excessive intake of the dosage form of the invention, the quantity of aversive agent consumed will exceed the "no effect" or "minimum effect" threshold, thereby producing one or more aversive effects, for example, e.g., nausea, emesis, diarrhea, laxation, cutaneous vasodilation, headache, bitter taste, naso-mucosal irritation, oro-mucosal irritation, reduction of the pleasurable, mood altering, rewarding, reinforcing, or other psychic and physiologic effects of the opioid agonist or a co-abused drug.
[00270] In some embodiments, the "no effect" or "minimum effect" threshold amount of the abuse intervention agent can be exceeded when the dosage form of the invention is taken in excess of the manufacturer's recommendation by a factor of about 1.5, or about 2, or about 2.5, or about 3, or about 4, or about 5, or about 6, or about 7, or about 8, or about 10, or more than 10. In some embodiments, the production of an aversive effect can reduce or stop further abuse of the dosage form, thereby reducing the harm or toxicity of the drug in the subject who is tampering, misusing or abusing the dosage form, e.g., addicts, drug abusers and recreational drug users.
Treatments
[00271] The opioid agonist of the invention may be used for the prevention or treatment of any diseases and disorders, including without limitation, (i) pain; (ii) infectious, immunologic, cardiovascular, pulmonary, gastrointestinal, hepatic, biliary, nutritional, metabolic, endocrine, hematologic, oncologic, musculoskeletal, rheumatic, neurologic, psychiatric, genitourinary, gynecologic, obstetric, pediatric, otolaryngogologic, ophthalmic, dermatologic, dental, oral, and genetic disorders, diseases and maladies and signs and symptoms thereof; (iii) depression, schizophrenia, influenza, common colds, anxiety, panic attacks, agoraphobia, ADHD, insomnia, sleep disorders, nasal congestion, headaches, migraine, urinary incontinence,
constipation, allergies, cough, pneumonia, COPD, asthma, fluid retention, acid reflux, peptic ulcers, hypertension, cardiac arrhythmias, hypercholesterolemia, CHF, fever, diarrhea, back pain, myofascial pain, osteoarthritis, neuropathic pain, cancer pain, acute pain, diabetes, muscle spasms, and rheumatoid arthritis, and signs and symptoms thereof; (iv) disorders, diseases and maladies, and signs and symptoms thereof referred to in Harrison's Principles of Internal Medicine, 16th Edition, 2004, Kasper DL, Braunwald W, Fauci A, Hauser S, Longo D, and Jameson JL (eds)]. The formulations of the invention may be used to treat painful conditions. As used herein, the term "pain" includes: (i) peripheral neuropathic pain, e.g., acute and chronic inflammatory demeyelinating polyradiculopathy, alcoholic polyneuropathy, chemotherapy-induced polyneuropathy, complex regional pain syndrome (CRPS) Type I and Type II, entrapment neuropathies (e.g., carpal tunnel syndrome), HIV sensory neuropathy, iatrogenic neuralgias (e.g., postthoracotomy pain, postmastectomy pain), idiopathic sensory neuropathy, painful diabetic neuropathy, phantom limb pain, postherpetic neuralgia, trigeminal neuralgia, radiculopathy (e.g., cervical thoracic, lumbosacral), sciatica, acute herpes zoster pain, temporomandibular joint disorder pain and postradiation plexopathy; and (ii) central neuropathic pain, e.g., compressive myelopathy from spinal stenosis, HFV myelopathy, multiple sclerosis pain, Parkinson's disease pain, postischemic myelopathy, post postradiation myelopathy, poststroke pain, posttraumatic spinal cord injury and syringomyelia; and (iii) cancer associated neuropathic pain, e.g., chemotherapy induced polyneuropathy, neuropathy secondary to tumor infiltration or nerve compression, phantom breast pain, postmastectomy pain, postradiation plexopathy and myelopathy; (iv) chronic pain, e.g., back pain, rheumatoid arthritis, osteoarthritis, inflammatory pain, non-inflammatory pain, myofascial pain, fibromyalgia, cancer pain, visceral pain, somatic pain, pelvic pain, musculoskeletal pain, post-traumatic pain, bone pain and idiopathic pain; (v) acute pain, e.g, acute postsurgical pain (including laparoscopic, laparatomy, gynecologic, urologic, cardiothoracic, arthroscopic, gastrointestinal, neurologic, orthopedic, oncologic, maxillofacial, ophthalmic,
otolaryngologic, soft tissue, plastic, cosmetic, vascular and podiatric surgery, including abdominal surgery, abdominoplasty, adenoidectomy, amputation, angioplasty, appendectomy, arthrodesis, arthroplasty, arthroscopy, bilateral cingulotomy, biopsy, brain surgery, breast biopsy, cauterization, cesarean section, cholecystectomy, circumcision, commissurotomy, cordotomy, corneal transplantation, cricothoracotomy, discectomy, diverticulectomy, episiotomy, endarterectomy, endoscopic thoracic sympathectomy, foreskin restoration, fistulotomy, frenectomy, frontalis lift, fundectomy, gastrectomy, grafting, heart transplantation, hemicorporectomy, hemorrhoidectomy, hepatectomy, hernia repair, hypnosurgery, hysterectomy, kidney transplantation, laminectomy, laparoscopy, laparotomy, laryngectomy, lithotripsy, lobotomy, lumpectomy, lung transplantation, mammectomy, mammoplasty, mastectomy, mastoidectomy, mentoplasty, myotomy, mryingotomy, nephrectomy, nissen fundoplication, oophorectomy, orchidectomy, parathyroidectomy, penectomy, phalloplasty, pneumotomy, pneumonectomy, prostatectomy, psychosurgery, radiosurgery, ritidoplasty, rotationplasty, sigmoidostomy, sphincterotomy, splenectomy, stapedectomy, thoracotomy, thrombectomy, thymectomy, thyroidectomy, tonsillectomy, tracheotomy, tracheostomy, tubal ligation, ulnar collateral ligament reconstruction, ureterosigmoidostomy, vaginectomy, vasectomy, vulvectomy; renal colic; incisional pain; inflammatory incisional pain; nociceptive incisional pain; acute neuropathic incisional pain following surgery), renal colic, trauma, acute back pain, burn pain, burn dressing change pain, migraine pain, tension headache pain, acute musculoskeletal pain, acute exacerbation or flare of chronic back pain, acute exacerbation or flare of osteoarthritis, acute exacerbation or flare of chronic pain, breakthrough chronic non-cancer pain, breakthrough cancer pain, acute exacerbation or flare of fibromylagia, acute exacerbation or flare of rheumatoid arthritis, acute exacerbation or flare of myofacsial pain, acute exacerbation or flare of chronic idiopathic pain, acute exacerbation or flare of neuropathic pain, procedure related pain (e.g., arthroscopy, laparoscopy, endoscopy, intubation, bone marrow biopsy, soft tissue biopsy, catheterization), and other self-limiting pain states.
[00273] Perceptible Pain Relief, Confirmed Perceptible Pain Relief and
Meaningful Pain Relief are assessed and defined as follows: At the time of dosing with the study medication, a trained member of study staff starts two stopwatches for each patient. The patient is instructed to stop the first stopwatch at the time of perceptible pain relief and the second stopwatch at the time when they first experience meaningful pain relief. The usual definitions of the perceptible and meaningful pain relief are as follows: Perceptible Pain Relief is when the patient begins to feel any pain relieving effect from the drug. The patient is typically instructed as follows: "I would like you to stop the first stopwatch when you first feel any pain relief whatsoever. This does not mean you feel completely better, although you might, but when you first feel any difference in the pain that you have had". Meaningful Pain Relief is when the patient feels their pain relief is meaningful to them. The patient is typically instructed as follows: "I would like you to stop the second stopwatch when you have meaningful pain relief. That is, when the relief from the pain is meaningful to you". Confirmed Perceptible Pain Relief is Perceptible Pain Relief in those patients who go on to also have Meaningful Pain Relief.
[00274] The "drug effects" questionnaire assesses the extent to which subjects currently felt a drug effect, on a scale of 1 to 5 (1 = "I feel no effect from it at all"; 2 = "I think I feel a mild effect, but I'm not sure"; 3 = "I feel an effect, but it is not real strong"; 4 = "I feel a strong effect"; 5 = "I feel a very strong effect"). This questionnaire can be used to examine the overall drug effects of abusable drugs given intact and upon tampering, preferably in drug abusers and recreational drug users without the medical condition for which the drug is effective.
[00275] The "drug liking" questionnaire assesses the extent to which subjects currently like the effects of the drug on a 100-mm VAS, bounded on the left by "0 = dislike a lot", bounded on the right by "100 = like a lot". This questionnaire can be used to examine the overall drug liking of abusable drugs given intact and upon tampering, preferably in drug abusers and recreational drug users without the medical condition for which the drug is effective.
[00276] The "take again" questionnaire assesses whether subjects would take the abusable drug again if given the opportunity. The patient is asked "If given an opportunity, would you take this drug again? (circle one: YES or NO). This questionnaire can be used to examine the overall desirability of the drug experience with the abusable drugs taken intact and taken after tampering, preferably in drug abusers and recreational drug users without the medical condition for which the drug is effective.
[00277] On the "coasting" questionnaire the patient is asked to put a mark on a horizontal line that best describes their response to the question: "Do you feel like you are coasting or spaced out? The horizontal line is a visual analog scale (VAS) bounded on the left by "not at all" and on the right by "extremely". This questionnaire can be used to examine the degree to which subjects feel like they are coasting or spaced out with the abusable drugs taken intact and taken after tampering, preferably in drug abusers and recreational drug users without the medical condition for which the drug is effective.
[00278] Three performance tasks may be employed for measuring skills related to driving.
[00279] The "critical tracking task" measures the patient's ability to control a displayed error signal in a first-order compensatory tracking task. The error is displayed as a horizontal deviation of a cursor from the midpoint on a horizontal, linear scale. Compensatory joystick movements correct the error by returning the cursor to the midpoint. The frequency at which the patient loses the control is the critical frequency. The critical tracking task measures the psychomotor control during a closed loop operation. It is a laboratory analog to on-the-road tracking performance.
[00280] The "stop signal task" measures motor impulsivity, which is defined as the inability to inhibit an activated or pre-cued response leading to errors of commission. The task requires patients to make quick key responses to visual go signals, i.e. the letters ABCD presented one at a time in the middle of the screen, and to inhibit any response when a visual stop signal, i.e. "*" in one of the four corners of the screen, is presented at predefined delays. The main
dependent variable is the stop reaction time on stop signal trials that represents the estimated mean time required to inhibit a response.
[00281] The Tower of London (TOL) is a decision-making task that measures executive function and planning. The task consists of computer generated images of begin- and end-arrangements of three colored balls on three sticks. The subject's task is to determine as quickly as possible, whether the end- arrangement can be accomplished by "moving" the balls in two to five steps from the beginning arrangement by pushing the corresponding number coded button. The total number of correct decisions is the main performance measure.
[00282] For the purposes of in vivo testing, unless specified otherwise, pain intensity is measured on a VAS or categorical scale. On the categorical scale, the patient is asked "My pain at this time is: None = 0, Mild = 1 , Moderate = 2, Severe = 3. On the VAS, the patient is asked "My pain at this time is" (with VAS anchors: "No Pain" and "Extreme Pain").
[00283] For the purposes of in vivo testing, unless specified otherwise, pain relief is measured on a categorical scale. The patient is asked "My relief from starting pain is: None = 0, A little = 1, Some = 2, A lot = 3, Complete = 4.
[00284] In certain preferred embodiments of the invention, wherein the opioid agonist is an analgesic and the sequestered agent is a cannabinoid antagonist, the mean ratio of the time to confirmed perceptible pain relief after administration of the tampered dosage form to the time to confirmed perceptible pain relief after administration of the intact dosage form is more than 20: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 15: 1, or more than about 10: 1, or more than about 7: 1, or more than about 5:1, or more than about 3:1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than 1.15: 1.
[00285] In certain preferred embodiments of the invention, wherein the opioid
■ agonist is an analgesic and the sequestered agent is a cannabinoid antagonist, the mean ratio of the time to meaningful pain relief after administration of the ' tampered dosage form to the time to meaningful pain relief after
administration of the intact dosage form is more than 20:1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than" about 15:1, or more than about 10: 1, or more than about 7: 1 , or more than about 5: 1 , or more than about 3 : 1 , or more than about 2: 1 , or more than about 1.5:1, or more than about 1.25:1, or more than 1.15: 1.
[00286] In certain preferred embodiments of the invention, wherein the opioid agonist is an analgesic and the sequestered agent is a cannabinoid antagonist, the mean ratio of the peak pain intensity difference score after administration of the intact dosage form to the peak pain intensity difference score after administration of the tampered dosage form is more than 10: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 8: 1, or more than about 7: 1, or more than about 5: 1, or more than about 3: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1.
(00287] In certain preferred embodiments of the invention, wherein the opioid agonist is an analgesic and the sequestered agent is a cannabinoid antagonist, the mean ratio of the peak pain relief score after administration of the intact dosage form to the peak pain relief score after administration of the tampered dosage form is more than 10: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 8: 1, or more than about 7: 1, or more than about 5: 1, or more than about 3: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15: 1.
[00288] In certain preferred embodiments of the invention, wherein the opioid agonist is an analgesic and the sequestered agent is a cannabinoid antagonist, the mean ratio of change from baseline to two hours post-dose or four hours post-dose in pain intensity score after administration of the intact dosage form to the change from baseline to two hours post-dose in pain intensity score after administration of the tampered dosage form is more than 10: 1; said pain score measured in acute postsurgical pan. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 9: 1, or more than about 8: 1, or more than about 7: 1, or more than about 5: 1, or more
than about 3:1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1 , or more than about 1.15: 1.
[00289] In certain preferred embodiments of the invention, wherein the opioid agonist is an analgesic and the sequestered agent is a cannabinoid antagonist, the mean ratio of the number of patients with pain who obtain 33% pain relief after administration of the intact dosage form when compared with following administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1.
[00290] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of the incidence and nausea intensity score in healthy subjects (previously naϊve to opioid agonist) after administration of the intact dosage form when compared with following administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2:1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 :1.
[00291] In certain preferred embodiments of the invention, wherein the sequestered agent is an alcohol deterrent or alcohol aversive agent, the mean ratio of the nausea intensity score in recreational drug users or drug addicts after administration of the tampered dosage form when compared with administration of the intact dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1: 1; said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00292] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of moderate or severe sedation or drowsiness in healthy subjects (naive to opioid agonist) after administration of the intact dosage form when compared with following administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15:1, or more than about 1 : 1. Preferably, the aforementioned sedation is measured at 0.5, 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
[00293] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of moderate or severe sedation or drowsiness in healthy subjects (naϊve to opioid agonist) after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 : 1; said dosage form administration followed about 0.5 hour later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00294] In certain preferred embodiments of the invention, wherein the sequestered agent is an alcohol deterrent or alcohol aversive agent, the mean ratio of incidence of moderate to severe headache in recreational drug users or drug addicts after administration of the tampered dosage form when compared with administration of the intact dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, said dosage form administration preceded
by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00295] In certain preferred embodiments of the invention, wherein the sequestered agent is an alcohol deterrent or alcohol aversive agent, the mean ratio of the incidence of vomiting or retching in recreational drug users or drug addicts after administration of the tampered dosage form when compared with administration of the intact dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00296] In certain preferred embodiments of the invention, wherein the sequestered agent is an alcohol deterrent or alcohol aversive agent, the mean ratio of the incidence of cutaneous flushing in recreational drug users or drug addicts after administration of the tampered dosage form when compared with following administration of the intact dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, said dosage form administration preceded^ by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00297] In certain preferred embodiments of the invention, wherein the sequestered agent is an alcohol deterrent or alcohol aversive agent, the mean ratio of the incidence of self-reported signs and symptoms selected from the group comprising skin flushing, throbbing, headache, difficulty breathing, nausea, vomiting, retching, sweating, thirst, chest pain, dizziness, fainting, anxiety, uneasiness, weakness, blurred vision and confusion, in recreational drug users or drug addicts after administration of the tampered dosage form when compared with following administration of the intact dosage form is
more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2:1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15:1, said dosage form administration preceded by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00298] In certain preferred embodiments of the invention, the mean ratio of the drug liking score in drug abusers and recreational drug users after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1:1. Preferably, the aforementioned drug liking score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
[00299] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of the score on the "take again" questionnaire in drug abusers and recreational drug users after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 :1. Preferably, the aforementioned take again score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
[00300] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of the score on the "coasting" questionnaire in drug abusers and recreational drug users after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1. In other embodiments of the
invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2:1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15: 1, or more than about 1: 1. Preferably, the aforementioned coasting score is measured at 2, 3, 4, 5 or 6 hours, or up to 2, 3, 4, or 6 hours post-dose.
[00301J In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of impairment on the "critical tracking task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1. In other , embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25:1, or more than about 1.15:1, or more than about 1:1. Preferably, the aforementioned critical tracking task score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
[00302] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of impairment on the "critical tracking task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1: 1; said "critical tracking task" driving skills test score measured 2.5 to 6 hours after administration of the dosage form, said dosage form administration followed about 1 hours later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
[00303] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of impairment on the "stop signal task" driving skills test in opioid agonist naive healthy
subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15:1, or more than about 1 : 1. Preferably, the aforementioned stop signal task score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
[00304] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of impairment on the "stop signal task" driving skills test in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3: 1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5: 1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1 : 1; said "critical tracking task" driving skills test score measured 2.5 to 6 hours after administration of the dosage form, said dosage form administration followed about 1 hours later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
|00305j In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of impairment on the "Tower of London" driving skills test score in opioid agonist naϊve healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5:1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4:1, or more than about 3:1, or more than about 2.5: 1, or more than about 2: 1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1; or more than about 1 : 1. Preferably, the aforementioned Tower of London score is measured at 1, 1.5, 2, 3, 4, 5 or 6 hours, or up to 1, 2, 3, 4, or 6 hours post-dose.
[00306] In certain preferred embodiments of the invention, wherein the sequestered agent is a cannabinoid antagonist, the mean ratio of impairment on the "Tower of London" driving skills test score in opioid agonist naive healthy subjects after administration of the intact dosage form when compared with administration of the tampered dosage form is more than 5: 1. In other embodiments of the invention, the mean ratio using the aforementioned test method is more than about 4: 1, or more than about 3: 1, or more than about 2.5:1, or more than about 2:1, or more than about 1.5:1, or more than about 1.25: 1, or more than about 1.15: 1, or more than about 1:1; said "critical tracking task" driving skills test score measured 2.5 to 6 hours after administration of the dosage form, said dosage form administration followed about 1 hours later by alcohol (ethanol) administration sufficient to maintain a blood alcohol concentration of 0.04% to 0.08%.
Preparation of Releasable Opioid agonist as an Immediate Release Dosage Form [00307] Pharmaceutical composition and methods of the present invention comprise one or more opioid agonists in releasable form and one or more aversive agents in sequestered (i.e., non-releasable or substantially releasable) form chosen from the group comprising cannabinoid antagonists, and alcohol deterrents, and combinations thereof. In certain preferred embodiments of the invention, the dosage form optionally comprises, in addition to the foregoing, one of more abuse intervention agents in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form in unsalified form xenobiotics base or pharmaceutically acceptable salts in racemic or enantiomeric form, or mixtures thereof in and they are intended for oral administration.
[00308] All oral pharmaceutical dosage forms of the invention are contemplated, including oral suspensions, tablets, capsules, lozenges, effervescent tablets, effervescent powders, powders, solutions, powders for reconstitution, transmucosal films, buccal products, oral mucoretentive products, oral gastroretentive tablets and capsules, orally disintegrating tablets, fast dissolving tablets, fast dispersing tablets, fast disintegrating dosage forms, administered as immediate release, modified release, enteric coated, sustained
release, controlled release, pulsatile release, delayed release, colonic delivery, targeted delivery and extended release dosage form.
[00309] The preparation of oral immediate release dosage forms is well known in the art - see Remington: the science of Pharmacy Practice, 21st Edition, 2006, Lippincott, Williams & Wilkins, Baltimore, MD; Pharmaceutical Preformulation and Formulation: A Practical Guide from Candidate Drug Selection to Commercial Dosage Form. Gibson, M (ed). CRC Press, 2001; Niazi, S. Handbook of Pharmaceutical Manufacturing Formulations: Uncompressed Solid Products (Volume 2 of 6), CRC Press, 2004; Niazi, S. Handbook of Pharmaceutical Manufacturing Formulations: Compressed Solid Products (Volume 1 of 6), CRC Press, 2004; Mollet, H, Grubenmann A, Payne H. Formulation Technology: Emulsions, Suspensions, Solid Forms, Wiley- VCH, 2001; Niazi S and Niazi SK (all of which are hereby incorporated by reference). A majority of oral dosage forms commercially available world wide are formulated as immediate release products. A wide variety of immediate release dosage forms can be formulated, including oral suspensions, tablets, capsules, lozenges, effervescent tablets, effervescent powders, powders, solutions, powders for reconstitution.
Preparation of Aversive Agent (e.g., Cannabinoid Antagonist) in a Substantially Non-releasable Form
[00310] In certain embodiments of the present invention, the opioid agonist is in an immediate release form and the aversive agent is in a substantially non- releasable form. In certain embodiments of the present invention, an aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form may be prepared by combining the aversive agent (e.g., antagonist) with one or more of a pharmaceutically acceptable hydrophobic material. For example, aversive agent (e.g., cannabinoid antagonist) particles may be coated with coating that substantially prevents the release of the aversive agent (e..g., antagonist), the coating comprising the hydrophobic materials(s). Another example would be an aversive agent (e.g., cannabinoid antagonist) that is dispersed in a matrix that renders the aversive agent (e.g., antagonist) to be
substantially non-releasable, the matrix comprising the hydrophobic materials(s). In certain embodiments, the pharmaceutical acceptable hydrophobic material comprises a cellulose polymer selected from the group consisting of ethylcellulose, cellulose acetate, cellulose propionate (lower, medium or higher molecular weight), cellulose acetate propionate, cellulose acetate butyrate, cellulose acetate phthalate and cellulose triacetate. An example of ethylcellulose is one that has an ethoxy content of 44 to 55%. Ethylcellulose may be used in the form of an alcoholic solution. In certain other embodiments, the hydrophobic material comprises polylactic acid, polyglycolic acid or a co-polymer of the polylactic and polyglycolic acid.
[00311] In certain embodiments, the hydrophobic material may comprise a cellulose polymer selected from the group consisting of cellulose ether, cellulose ester, cellulose ester ether, and cellulose. The cellulosic polymers have a degree of substitution, D. S., on the anhydroglucose unit, from greater than zero and up to 3 inclusive. By degree of substitution is meant the average number of hydroxyl groups present on the anhydroglucose unit comprising the cellulose polymer that are replaced by a substituting group. Representative materials include a polymer selected from the group consisting of cellulose acylate, cellulose diacylate, cellulose triacylate, cellulose acetate, cellulose diacetate, cellulose triacetate, mono, di, and tricellulose alkanylates, mono, di, and tricellulose aroylates, and mono, di, and tricellulose alkenylates. Exemplary polymers include cellulose acetate having a D.S. and an acetyl content up to 21%; cellulose acetate having an acetyl content up to 32 to 39.8%; cellulose acetale having a D.S. of 1 to 2 and an acetyl content of 21 to 35%; cellulose acetate having a D.S. of 2 to 3 and an acetyl content of 35 to 44.8%.
[00312] More specific cellulosic polymers include cellulose propionate having a D.S. of 1.8 and a propyl content of 39.2 to 45 and a hydroxyl content of 2.8 to 5.4%;, cellulose acetate butyrate having a D.S. of 1.8, an acetyl content of 13 to 15% and a butyryl content of 34 to 39%; cellulose acetate butyrate having an acetyl content of 2 to 29%, a butyryl content of 17 to 53% and a hydroxyl content of 0.5 to 4.7%; cellulose triacylate having a D.S. of 2.9 to 3
such as cellulose triacetate, cellulose trivalerate, cellulose trilaurate, cellulose tripatmitate, cellulose trisuccinate, and cellulose trioctanoate; cellulose diacylates having a D. S. of 2.2 to 2.6 such as cellulose disuccinate, cellulose dipalmitate, cellulose dioctanoate, cellulose dipentanoate, and coesters of cellulose such as cellulose acetate butyrate, cellulose acetate octanoate butyrate and cellulose acetate propionate.
[00313] Additional cellulose polymers useful for preparing an aversive agent
(e.g., cannabinoid antagonist) in a substantially non-releasable form includes acetaldehyde dimethyl cellulose acetate, cellulose acetate ethylcarbamate, cellulose acetate methylcarbamate, and cellulose acetate dimethylaminocellulose acetate.
[00314] An acrylic polymer useful for preparation of the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form includes, but are not limited to, acrylic resins comprising copolymers synthesized from acrylic and methacrylic acid esters (e.g., the copolymer of acrylic acid lower alkyl ester and methacrylic acid lower alkyl ester) containing about 0.02 to 0.03 mole of a tri (lower alkyl) ammonium group per mole of the acrylic and methacrylic monomers used. An example of a suitable acrylic resin is Eudragit RS. Eudragit RS30D is preferred. Eudragit RS is a water insoluble copolymer of ethyl acrylate (EA), methyl methacrylate (MM) and trimethylammoniumethyl methacrylate chloride (TAM) in which the molar ratio of TAM to the remaining components (EA and MM) is 1 :40. Acrylic resins such as Eudragit RS may be used in the form of an aqueous suspension.
[00315] In certain embodiments of the invention, the acrylic polymer may be selected from the group consisting of acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamide copolymer, poly(methyl methacrylate), polymethacrylate, poly(methyl methacrylate) copolymer, polyacrylamide, aminoalkyl methacrylate copolymer, poly(methacrylic acid anhydride), and glycidyl methacrylate co-polymers.
[00316] When the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form comprises aversive agent (e.g., cannabinoid antagonist) particles coated with a coating that renders the aversive agent (e.g., antagonist) substantially non-releasable, and when a cellulose polymer or an acrylic polymer is used for preparation of the coating composition, suitable plasticizers, e.g., acetyl triethyl citrate and/or acetyl tributyl citrate may also be admixed with the polymer. The coating may also contain additives such as coloring agents, talc and/or magnesium stearate, which are well known in the coating art.
[00317] The coating composition may be applied onto the aversive agent (e.g., cannabinoid antagonist) particles by spraying it onto the particles using any suitable spray equipment known in the part. For example, a Wuster fluidized- bed system may be used in which an air jet, injected from underneath, fluidizes the coated material and effects drying while the insoluble polymer coating is sprayed on. The thickness of the coating will depend on the characteristics of the particular coating composition being used. However, it is well within the ability of one skilled in the art to determine by routine experimentation the optimum thickness of a particular coating required for a particular dosage form of the present invention.
[00318] The pharmaceutically acceptable hydrophobic material useful for preparing a cannabinoid antagonist in a substantially non-releasable form includes a biodegradable polymer comprising a poly(lactic/glycolic acid) ("PLGA"), a polylactide, a polyglycolide, a polyanhydride, a polyorthoester, polycaprolactones, polyphosphazenes, polysaccharides, proteinaceous polymers, polyesthers, polydioxanone, polygluconate, polylactic-acid- polyethylene oxide copolymers, poly(hydroxybutyrate), polyphosphoesther or mixtures or blends of any of these.
[00319] In certain embodiments, biodegradable polymer comprises a poly(lactic/glycolic acid), a copolymer of lactic and glycolic acid, having molecular weight of about 2,000 to about 500,000 daltons. The ratio of lactic acid to glycolic acid is from about 100:0 to about 25:75, with the ratio of lactic acid to glycolic acid of 65:35 being preferred.
[00320] Poly(lactic/glycolic acid) may be prepared by the procedure set forth in
U.S. Pat. No. 4,293,539 (Ludwig et al.), the disclosure of which is hereby incorporated by reference in its entirety. In brief, Ludwig prepares the copolymer by condensation of lactic acid and glycolic acid in the presence of a readily removable polymerization catalyst (e.g., a strong acid ion-exchange resin such as Dowex HCR-W2-H). The amount of catalyst is not critical to the polymerization, but typically is from about 0.01 to about 20 parts by weight relative to the total weight of combined lactic acid and glycolic acid. The polymerization reaction may be conducted without solvents at a temperature from about 100 C. to about 250 C. for about 48 to about 96 hours, preferably under a reduced pressure to facilitate removal of water and by-products. Poly(lactic/glycolic acid) is then recovered by filtering the molten reaction mixture in an organic solvent such as dichloromethane or acetone and then filtering to remove the catalyst.
[00321] Once the aversive agent (e.g., cannabinoid antagonist) in a substantially non-releasable form is prepared, it may be combined with an opioid agonist, along with conventional excipients known in the art, to prepare the oral dosage form of the present invention.
[00322] In certain preferred embodiments of the invention, the oral dosage form is a capsule or a tablet. When being formulated as a tablet, the aversive agent (e.g., cannabinoid antagonist) and agonist may be combined with one or more inert, non-toxic pharmaceutical excipients which are suitable for the manufacture of tablets. Such excipients include, for example, an inert diluent such as lactose; granulating and disintegrating agents such as cornstarch; binding agents such as starch; and lubricating agents such as magnesium stearate.
[00323] The oral dosage form of the present invention may be formulated to provide immediate release of the opioid agonist contained therein. In other embodiments of the invention, however, the oral dosage form provides sustained-release of the opioid agonist.
[00324] In certain embodiments, the oral dosage forms providing sustained release of the opioid agonist may be prepared by admixing the aversive agent
(e.g., cannabinoid antagonist) in a substantially non-releasable form with the agonist and desirable pharmaceutical excipients to provide a tablet, and then coating the tablet with a sustained-release tablet coating.
[00325] In certain embodiments of the invention, sustained release opioid agonist tablets may be prepared by admixing the substantially non-releasable form of an aversive agent (e.g., cannabinoid antagonist) with an aversive agent (e.g., cannabinoid antagonist) in a matrix that provides the tablets with sustained-releasing properties.
[00326] Detailed description for preparing sustained-release oral dosage forms according to the present invention is set forth below.
[00327] In some embodiments, the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 6 hours comprising (i) an aversive agent in releasable form and (ii) a substantially non-releasable aversive agent.
[00328] In some embodiments, the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 8 hours comprising (i) an opioid agonist in releasable form and (ii) a substantially non-releasable aversive agent.
[00329] In some embodiments, the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 12 hours comprising (i) an opioid agonist in releasable form and (ii) a substantially non-releasable aversive agent.
[00330] In some embodiments, the present invention is directed to oral dosage forms with an intended therapeutic effect of up to about 24 hours comprising (i) an opioid agonist in releasable form and (ii) a substantially non-releasable aversive agent.
Preparation of Controlled Release Dosage Forms Containing an Opioid Agonist and a Substantially Non-Releasable Form of an Aversive Agent
[00331] A combination of the opioid agonist and a substantially non-releasable form of an aversive agent (e.g., cannabinoid antagonist) may be formulated as a controlled or sustained release oral formulation in any suitable tablet, coated tablet or multiparticulate formulation known to those skilled in the art. The
sustained release dosage form may optionally include a sustained release carrier which is incorporated into a matrix along with the opioid agonist and a non-available form of an aversive agent (e.g., cannabinoid antagonist), or may be applied as a sustained release coating.
[00332] In one preferred embodiment of the present invention, the sustained release dosage form comprises such particles comprising the opioid agonist, wherein the particles have diameter from about 0.1 mm to about 2.5 mm, preferably from about 0.5 mm to about 2 mm.
[00333] The opioid agonist particles are preferably film coated with a material that permits release of the opioid agonist at a sustained rate in an aqueous medium. The film coat is chosen so as to achieve, in combination with the other stated properties, a desired in-vitro release rate. The sustained release coating formulations of the present invention should be capable of producing a strong, continuous film that is smooth and elegant, capable of supporting pigments and other coating additives, non-toxic and inert.
[00334] The dosage forms comprising an opioid agonist and a substantially non-releasable aversive agent (e.g., cannabinoid antagonist) may optionally be coated with one or more materials suitable for the modulation of the opioid agonist release or for the protection of the formulation. In one embodiment, coatings are provided to permit either pH-dependent or pH-independent release, e.g., when exposed to gastrointestinal fluid. A pH-dependent coating serves to release the cannabinoid in desired areas of the gastro-intestinal (GI) tract, e.g., the stomach or small intestine, such that an absorption profile is provided which is capable of providing at least about six hours and preferably about twelve hours to up to about twenty-four hours of therapeutic effect to a patient. When a pH-independent coating is desired, the coating is designed to achieve optimal release of the cannabinoid regardless of pH-changes in the environmental fluid, e.g., the GI tract. It is also possible to formulate compositions which release a portion of the dose in one desired area of the GI tract, e.g., the stomach, and release the remainder of the dose in another area of the GI tract, e.g., the small intestine.
[00335] Formulations according to the invention that utilize pH-dependent coatings to obtain formulations may also impart a repeat-action effect whereby unprotected drug is coated over the enteric coat and is released in the stomach, while the remainder, being protected by the enteric coating,-is released further down the gastrointestinal tract. Coatings which are pH-dependent may be used in accordance with the present invention include shellac, cellulose acetate phthalate (CAP), polyvinyl acetate phthalate (PVAP), hydroxypropyl methylcellulose phthalate, and methacrylic acid ester copolymers, zein, and the like.
(00336] In certain preferred embodiments, the substrate (e.g., tablet core bead, matrix particle) containing the cannabinoid analgesic is coated with a hydrophobic material selected from (i) an alkylcellulose; (ii) an acrylic polymer; or (iii) mixtures thereof. The coating may be applied in the form of an organic or aqueous solution or dispersion. The coating may be applied to obtain a weight gain from about 2 to about 25% of the substrate in order to obtain a desired sustained release profile.
[00337] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists, and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours. In other preferred embodiments, the dosage form provides said an in-vitro release rate of from 0% to about 40% at 1 hour, from about 5% to about 55% at 2 hours, from about 10% to about 60% at 4 hours, from about 15% to about 70% at 6 hours,
from about 25% to about 80% at 9 hours, and greater than about 50% at 12 hours.
[00338] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
[00339J In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours; said in-vitro release rate being substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
[00340] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for once-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours.
[00341] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for once-a-day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours; said in-vitro release rate being substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
[00342] In some preferred embodiments, the opioid agonist dosage forms provide an in-vitro release of from 2% to about 50% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at
one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C.
[00343] In some preferred embodiments, the opioid agonist dosage form provides an in-vitro release from about 5% to about 45% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C.
[00344) In some preferred embodiments, the opioid agonist dosage form provides a Cmax of opioid agonist which is less than 65% of the Cmax of an equivalent dose of an oral immediate release opioid agonist solution.
[00345] In some preferred embodiments, the dosage form provides a time to
75% mean Cmax of opioid agonist which is about 100% to about 2000% of the time to 75% mean Cmax of an oral immediate release opioid agonist solution.
[00346] In some preferred embodiments, the dosage from maintains a plasman opioid agonist concentration within 50% of Cmax for about 1 to about 9 hours during the 12 hour dosing interval.
[00347] In some preferred embodiments, the dosage from maintains a plasman opioid agonist concentration within 50% of Cmax for about 1 to about 9 hours during the 24 hour dosing interval.
[00348] In some preferred embodiments, the dosage form provides a Tmax of opioid agonist at a time point 1.5 to 20 times later than the Tmaχ provided by an equivalent dose of an immediate release opioid agonist solution. In the dosage form provides a Tmax at a time point about 1.5 to 15 times late, or about of 1.5 to 10 times later, or about of 1.5 to 7 times later, or about of 1.5 to 3 times later, or about of 2 to 20 times later, or about of 2 to 10 times later, or about of 2 to 5 times later, or about of 2 to 3 times later, or about of 2.5 to 20 times later, or about of 2.5 to 8 times later, or about of 2.5 to 5 times later, or about of 2.5 to 4 times later, or about of 3 to 20 times later, or about of 3 to 10 times later, or about of 3 to 5 times later.
[00349] In some preferred embodiments, the dosage form provides a mean in vivo extent of absorption of opioid agonist from 0 to 4 hours which is at least 20% of the mean in vivo extent of absorption from to 0 to 12 hours, wherein
the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve from the time of drug administration to the specified time point. In other embodiments, said in vivo extent of absorption from 0 to 4 hours is at least about 5%, or at least about 10%, or at least about 15%, or at least about 25%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, at least about 90%, or about 100% of the mean in vivo extent of absorption from to 0 to 12 hours.
[00350] In some preferred embodiments, the dosage form provides a mean in vivo extent of absorption of opioid agonist from 0 to 8 hours which is at least 20% of the mean in vivo extent of absorption from to 0 to 24 hours, wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve from the time of drug administration to the specified time point. In other embodiments, said in vivo extent of absorption from 0 to 8 hours is at least about 5%, or at least about 10%, or at least about 15%, or at least about 25%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, at least about 90%, or about 100% of the mean in vivo extent of absorption from to 0 to 24 hours.
[00351] In some preferred embodiments, the dosage form provides a mean in vivo extent of absorption of opioid agonist from 0 to 12 hours which is at least 20% of the mean in vivo extent of absorption from to 0 to 24 hours, wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve from the time of drug administration to the specified time point. In other embodiments, said in vivo extent of absorption from 0 to 12 hours is at least about 5%, or at least about 10%, or at least about 15%, or at least about 25%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, at least about 90%, or about 100% of the mean in vivo extent of absorption from to 0 to 24 hours.
[00352] In some preferred embodiments, the dosage form provides a mean in vivo extent of absorption of opioid agonist over the dosing interval (e.g., from
0 to 12 hours or from 0 to 24 hours) which is at least 40% of the mean in vivo extent of absorption from to 0 to ∞, wherein the mean in vivo extent of absorption is the area under the plasma or serum opioid agonist concentration time curve (AUC) from the time of drug administration to the specified time point and where AUC infinity is the sum of AUC from time "0" to time "t" (the last quantifiable time point which has been sampled) plus the extrapolated AUC from the last quantifiable sampling time point to infinity.
[00353] In some preferred embodiments, the dosage form provides an oral pharmaceutical composition of opioid agonist or a pharmaceutically acceptable salt thereof or mixtures thereof, said dosage form providing an in- vitro release of between 0% to about 50% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C.
[00354] In some preferred embodiments, the dosage form provides an oral pharmaceutical composition of opioid agonist or a pharmaceutically acceptable salt thereof or mixtures thereof, said dosage form providing an in- vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% to about 80% at 0.5 hours, and greater than about 40% at 1 hour; or between 0% to about 90% at 0.5 hours, and greater than about 60% at 1 hour; or between 1.6 and 7.2 at 37 0C of between 0% to about 100% at 0.5 hours, and greater than about 60% at 1 hour.
[00355] In some preferred embodiments, the dosage form provides an oral pharmaceutical composition of opioid agonist or a pharmaceutically acceptable salt thereof or mixtures thereof, said dosage form providing an in- vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% to about 90% at 1 hour, and greater than about 40% at 2 hours; or between 1.6 and 7.2 at 37 0C of between 0% to about 90% at 1 hour, and greater than about 70% at 2 hours; or between 1.6 and 7.2 at 37 0C of between 0% to about 50% at 1 hour, and greater than
about 30% at 2 hours; between 1.6 and 7.2 at 37 0C of between 0% to about 30% at 1 hour, and greater than about 25% at 2 hours; or between 1.6 and 7.2 at 37 0C of between 0% to about 100% at 1 hour, and greater than about 60% at 2 hours.
[00356] In some preferred embodiments, the invention comprises an oral pharmaceutical dosage form providing an in-vitro release of between 0% to about 50% by weight of the opioid agonist or a pharmaceutically acceptable salt thereof from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C.
[00357] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 3% and about 95% at 4 hours and between about 10% and about 100% at 8 hours; or between 10% and about 65% at 1 hour, between about 20% and about 75% at 2 hours, between about 30% and about 95% at 4 hours and between about 40% and about 100% at 8 hours.
[00358] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH
of between 1.6 and 7.2 at 37 0C of between 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
[00359] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 5% and about 50% at 1 hour, between about 10% and about 75% at 2 hours, between about 20% and about 95% at 4 hours, between about 40% and about 100% at 8 hours, greater than about 50% at 12 hours, greater than about 70% at 18 hours, and greater than about 80% at 24 hours; or between 5% and about 50% at 1 hour, between about 10% and about 75% at 2 hours, between about 20% and about 95% at 4 hours, between about 40% and about 100% at 8 hours, greater than about 50% at 12 hours, greater than about 70% at 18 hours, and greater than about 80% at 24 hours; or between 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours; or between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 25% and about 100% at 12 hours, between about 30% and about 100% at 16 hours, between about 50% and about 100% at 24 hours, and greater than about 80% at 36 hours.
[00360] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive
agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 20% and about 50% at 1 hour, between about 40% and about 75% at 2 hours, between about 60% and about 95% at 4 hours, between about 80% and about 100% at 8 hours and between about 90% and about 100% at 12 hours.
[00361J In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 10% and about 95% at 4 hours, between about 35% and about 100% at 8 hours, between about 55% and about 100% at 12 hours, between about 70% to about 100% at 16 hours, and greater than about 90% at 24 hours.
[00362] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 30% at 1 hour, between about 0% and about 45% at 2 hours, between about 3% and about
55% at 4 hours, between about 10% and about 65% at 8 hours, between about 20% and about 75% at 12 hours, between about 30% to about 88% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours.
[00363] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 20% and about 100% at 12 hours, between about 30% to about 100% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours.
[00364] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 15% and about 25% at 1 hour, between about 25% and about 35% at 2 hours, between about 30% and about 45% at 4 hours, between about 40% and about 60% at 8 hours, between about 55% and about 70% at 12 hours and between about 60% to about 75% at 16 hours.
[00365] In some preferred embodiments, the dosage form comprises a therapeutically effective amount of an opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, one or more sequestered aversive agents selected from the group comprising cannabinoid antagonists and alcohol deterrents, and controlled release material to render said dosage form suitable for twice-a-day administration to a human patient; said dosage form providing an in-vitro release rate of opioid agonist which is substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
[00366] In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C of between 0% to about 50% by weight of the opioid agonist. In other preferred embodiments, said release rate is between 0% to about 1%, or 0% to about 3%, or 0% to about 5%, or 0% to about 10%, or 0% to about 15%, or 0% to about 20%, 0% to about 30%, or 0% to about 40%, or 0% to about 60%, or 0% to about 70%, or 0% to about 80%, or 0% to about 90%, 0% to about 100%.
[00367] In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 3% and about 95% at 4 hours and between about 10%
and about 100% at 8 hours. In other preferred embodiments, said release rate is between 0% and about 10% at 1 hour, between about 0% and about 20% at 2 hours, between about 2% and about 80% at 4 hours and between about 5% and about 100% at 8 hours; or between 0% and about 20% at 1 hour, between about 0% and about 40% at 2 hours, between about 0% and about 80% at 4 hours and between about 2% and about 100% at 8 hours; or between 0% and about 40% at 1 hour, between about 0% and about 60% at 2 hours, between about 5% and about 85% at 4 hours and between about 5% and about 90% at 8 hours and greater than 20% at 12 hours; or between 0% and about 50% at 1 hour, between about 0% and about 50% at 2 hours, between about 10% and about 90% at 4 hours and between about 15% and about 90% at 8 hours and greater than 30% at 12 hours; or between 0% and about 70% at 1 hour, between about 0% and about 70% at 2 hours, between about 10% and about 75% at 4 hours and between about 15% and about 90% at 8 hours and greater than 30% at 12 hours. J In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 10% and about 65% at 1 hour, between about 20% and about 75% at 2 hours, between about 30% and about 95% at 4 hours and between about 40% and about 100% at 8 hours. In other preferred embodiments, said release rate is between 2% and about 70% at 1 hour, between about 5% and about 80% at 2 hours, between about 10% and about 90% at 4 hours and between about 20% and about 100% at 8 hours; or between 5% and about 60% at 1 hour, between about 10% and about 75% at 2 hours, between about 15% and about 85% at 4 hours and between about 30% and about 100% at 8 hours; or between 20% and about 70% at 1 hour, between about 20% and about 75% at 2 hours, between about 20% and about 90% at 4 hours and between about 40% and about 100% at 8 hours; or between 30% and about 80% at 1 hour,
between about 40% and about 85% at 2 hours, between about 40% and about 90% at 4 hours and between about 60% and about 100% at 8 hours; or between 1% and about 20% at 1 hour, between about 5% and about 20% at 2 hours, between about 10% and about 40% at 4 hours and between about 20% and about 40% at 8 hours and greater than 40% at 12 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours. In other preferred embodiments, said release rate is between 0% to about 30% at 1 hour, from about 5% to about 45% at 2 hours, from about 10% to about 60% at 4 hours, from about 15% to about 70% at 6 hours, from about 25% to about 80% at 9 hours, and greater than about 50% at 12 hours; or between 0% to about 20% at 1 hour, from about 2% to about 35% at 2 hours, from about 5% to about 50% at 4 hours, from about 10% to about 60% at 6 hours, from about 15% to about 70% at 9 hours, and greater than about 40% at 12 hours; or between 0% to about 10% at 1 hour, from about 1% to about 30% at 2 hours, from about 5% to about 40% at 4 hours, from about 10% to about 60% at 6 hours, from about 15% to about 70% at 9 hours, and greater than about 40% at 12 hours; or between 0% to about 5% at 1 hour, from about 0% to about 10% at 2 hours, from about 2% to about 20% at 4 hours, from about 5% to about 30% at 6 hours, from about 10% to about 40% at 9 hours, and greater than about 30% at 12 hours; or between 0% to about 50% at 1 hour, from about 15% to about 70% at 2 hours, from about 20% to about 75% at 4 hours, from about 30% to about 80% at 6 hours, from about 30% to about 90% at 9 hours, and greater than about 70% at 12 hours; or between 0% to about 60% at 1 hour, from about 15% to about
80% at 2 hours, from about 25% to about 85% at 4 hours, from about 35% to about 90% at 6 hours, from about 40% to about 90% at 9 hours, and greater than about 80% at 12 hours; ; or between 0% to about 70% at 1 hour, from about 20% to about 80% at 2 hours, from about 25% to about 80% at 4 hours, from about 35% to about 80% at 6 hours, from about 40% to about 80% at 9 hours, and greater than about 60% at 12 hours; or between 0% to about 75% at 1 hour, from about 30% to about 80% at 2 hours, from about 35% to about 90% at 4 hours, from about 50% to about 90% at 6 hours, from about 55% to about 95% at 9 hours, and greater than about 70% at 12 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 5% and about 50% at 1 hour, between about 10% and about 75% at 2 hours, between about 20% and about 95% at 4 hours, between about 40% and about 100% at 8 hours, greater than about 50% at 12 hours, greater than about 70% at 18 hours, and greater than about 80% at 24 hours. In other preferred embodiments, said release rate is between 2% and about 50% at 1 hour, between about 5% and about 75% at 2 hours, between about 15% and about 75% at 4 hours, between about 30% and about 90% at 8 hours, greater than about 40% at 12 hours, greater than about 60% at 18 hours, and greater than about 70% at 24 hours; or between 1% and about 40% at 1 hour, between about 2% and about 60% at 2 hours, between about 10% and about 65% at 4 hours, between about 20% and about 80% at 8 hours, greater than about 30% at 12 hours, greater than about 40% at 18 hours, and greater than about 60% at 24 hours; or between 5% and about 60% at 1 hour, between about 15% and about 80% at 2 hours, between about 25% and about 95% at 4 hours, between about 45% and about 100% at 8 hours, greater than about 60% at 12 hours, greater than about 80% at 18 hours, and greater than about 90% at 24 hours; or between 10% and about 65% at 1 hour, between about 20% and
about 85% at 2 hours, between about 30% and about 100% at 4 hours, between about 60% and about 100% at 8 hours, greater than about 70% at 12 hours, greater than about 90% at 18 hours, and greater than about 95% at 24 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours. In other preferred embodiments, said release rate is between 0% to about 20% at 1 hour, from about 5% to about 50% at 4 hours, from about 10% to about 60% at 8 hours, from about 15% to about 70% at 12 hours, from about 25% to about 90% at 18 hours, and greater than about 55% at 24 hours; or between 0% to about 10% at 1 hour, from about 5% to about 40% at 4 hours, from about 8% to about 50% at 8 hours, from about 10% to about 60% at 12 hours, from about 22% to about 80% at 18 hours, and greater than about 45% at 24 hours; or between 0% to about 35% at 1 hour, from about 15% to about 70% at 4 hours, from about 25% to about 75% at 8 hours, from about 30% to about 85% at 12 hours, from about 40% to about 100% at 18 hours, and greater than about 75% at 24 hours; or between 0% to about 40% at 1 hour, from about 20% to about 70% at 4 hours, from about 30% to about 80% at 8 hours, from about 35% to about 90% at 12 hours, from about 45% to about 100% at 18 hours, and greater than about 80% at 24 hours; or between 0% to about 45% at 1 hour, from about 25% to about 75% at 4 hours, from about 35% to about 85% at 8 hours, from about 40% to about 90% at 12 hours, from about 50% to about 100% at 18 hours, and greater than about 90% at 24 hours; or between 0% to about 50% at 1 hour, from about 30% to about 80% at 4 hours, from about 40% to about 90% at 8 hours, from about 45% to
about 95% at 12 hours, from about 60% to about 100% at 18 hours, and greater than about 95% at 24 hours; or between 0% to about 60% at 1 hour, from about 40% to about 80% at 4 hours, from about 45% to about 90% at 8 hours, from about 50% to about 100% at 12 hours, from about 70% to about 100% at 18 hours, and greater than about 80% at 24 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in- vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 25% and about 100% at 12 hours, between about 30% and about 100% at 16 hours, between about 50% and about 100% at 24 hours, and greater than about 80% at 36 hours. In other preferred embodiments, said release rate is between 0% and about 40% at 1 hour, between about 0% and about 65% at 2 hours, between about 2% and about 85% at 4 hours, between about 8% and about 90% at 8 hours, between about 20% and about 95% at 12 hours, between about 25% and about 95% at 16 hours, between about 40% and about 90% at 24 hours, and greater than about 70% at 36 hours; or between 0% and about 30% at 1 hour, between about 0% and about 50% at 2 hours, between about 1% and about 75% at 4 hours, between about 5% and about 80% at 8 hours, between about 10% and about 85% at 12 hours, between about 15% and about 90% at 16 hours, between about 30% and about 80% at 24 hours, and greater than about 70% at 36 hours; or between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 5% and about 100% at 4 hours, between about 15% and about 100% at 8 hours, between about 35% and about 100% at 12 hours, between about 40% and about 100% at 16 hours, between about 60% and about 100% at 24 hours, and greater than about 85% at 36 hours; or between 0% and about 65% at 1 hour, between about 0% and about 85% at 2
hours, between about 10% and about 100% at 4 hours, between about 20% and about 100% at 8 hours, between about 40% and about 100% at 12 hours, between about 50% and about 100% at 16 hours, between about 70% and about 100% at 24 hours, and greater than about 90% at 36 hours; or between 0% and about 70% at 1 hour, between about 0% and about 90% at 2 hours, between about 20% and about 100% at 4 hours, between about 30% and about 100% at 8 hours, between about 50% and about 100% at 12 hours, between about 60% and about 100% at 16 hours, between about 80% and about 100% at 24 hours, and greater than about 95% at 36 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 20% and about 50% at 1 hour, between about 40% and about 75% at 2 hours, between about 60% and about 95% at 4 hours, between about 80% and about 100% at 8 hours and between about 90% and about 100% at 12 hours. In other preferred embodiments, said release rate is between 15% and about 45% at 1 hour, between about 35% and about 70% at 2 hours, between about 55% and about 90% at 4 hours, between about 75% and about 90% at 8 hours and between about 80% and about 95% at 12 hours; or between 10% and about 40% at 1 hour, between about 30% and about 65% at 2 hours, between about 50% and about 85% at 4 hours, between about 70% and about 85% at 8 hours and between about 75% and about 90% at 12 hours; or between 5% and about 35% at 1 hour, between about 25% and about 60% at 2 hours, between about 45% and about 80% at 4 hours, between about 65% and about 80% at 8 hours and between about 70% and about 85% at 12 hours; or between 25% and about 55% at 1 hour, between about 45% and about 80% at 2 hours, between about 65% and about 95% at 4 hours, between about 85% and about 100% at 8 hours and between about 95% and about 100% at 12 hours; or between 30% and about 60% at 1 hour, between about 50% and
about 80% at 2 hours, between about 70% and about 95% at 4 hours, between about 90% and about 100% at 8 hours and between about 95% and about 100% at 12 hours; or between 35% and about 60% at 1 hour, between about 50% and about 80% at 2 hours, between about 80% and about 95% at 4 hours, between about 90% and about 100% at 8 hours and between about 95% and about 100% at 12 hours; or between 20% and about 40% at 1 hour, between about 40% and about 65% at 2 hours, between about 60% and about 85% at 4 hours, between about 70% and about 90% at 8 hours and between about 80% and about 100% at 12 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 10% and about 95% at 4 hours, between about 35% and about 100% at 8 hours, between about 55% and about 100% at 12 hours, between about 70% to about 100% at 16 hours, and greater than about 90% at 24 hours. In other preferred embodiments, said release rate is between 0% and about 40% at 1 hour, between about 0% and about 65% at 2 hours, between about 8% and about 85% at 4 hours, between about 30% and about 90% at 8 hours, between about 45% and about 100% at 12 hours, between about 60% to about 100% at 16 hours, and greater than about 80% at 24 hours; or between 0% and about 30% at 1 hour, between about 0% and about 55% at 2 hours, between about 5% and about 75% at 4 hours, between about 20% and about 80% at 8 hours, between about 35% and about 100% at 12 hours, between about 50% to about 100% at 16 hours, and greater than about 70% at 24 hours; or between 0% and about 20% at 1 hour, between about 0% and about 45% at 2 hours, between about 5% and about 65% at 4 hours, between about 10% and about 70% at 8 hours, between about 25% and about 80% at 12 hours, between about 40% to about 100% at 16 hours, and
greater than about 60% at 24 hours; or between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 15% and about 95% at 4 hours, between about 40% and about 100% at 8 hours, between about 60% and about 100% at 12 hours, between about 75% to about 100% at 16 hours, and greater than about 90% at 24 hours; or between 0% and about 65% at 1 hour, between about 0% and about 85% at 2 hours, between about 20% and about 90% at 4 hours, between about 45% and about 100% at 8 hours, between about 65% and about 100% at 12 hours, between about 80% to about 100% at 16 hours, and greater than about 90% at 24 hours; or between 0% and about 40% at 1 hour, between about 0% and about 50% at 2 hours, between about 10% and about 80% at 4 hours, between about 25% and about 70% at 8 hours, between about 40% and about 80% at 12 hours, between about 60% to about 100% at 16 hours, and greater than about 90% at 24 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 30% at 1 hour, between about 0% and about 45% at 2 hours, between about 3% and about 55% at 4 hours, between about 10% and about 65% at 8 hours, between about 20% and about 75% at 12 hours, between about 30% to about 88% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours. In other preferred embodiments, said release rate is between 0% and about 25% at 1 hour, between about 0% and about 40% at 2 hours, between about 2% and about 50% at 4 hours, between about 8% and about 60% at 8 hours, between about 10% and about 70% at 12 hours, between about 25% to about 80% at 16 hours, between about 45% and about 100% hours at 24 hours and greater than 75% at 36 hours; or between 0% and about 20% at 1 hour, between about 0% and about 35% at 2 hours, between about 1% and about 45% at 4 hours, between about 5% and about 55% at 8 hours, between about 8% and about
65% at 12 hours, between about 20% to about 75% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 15% at 1 hour, between about 0% and about 30% at 2 hours, between about 0% and about 40% at 4 hours, between about 5% and about 50% at 8 hours, between about 8% and about 60% at 12 hours, between about 15% to about 70% at 16 hours, between about 35% and about 100% hours at 24 hours and greater than 60% at 36 hours; or between 0% and about 10% at 1 hour, between about 0% and about 25% at 2 hours, between about 0% and about 35% at 4 hours, between about 5% and about 45% at 8 hours, between about 10% and about 50% at 12 hours, between about 10% to about 60% at 16 hours, between about 30% and about 90% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 35% at 1 hour, between about 0% and about 50% at 2 hours, between about 5% and about 60% at 4 hours, between about 15% and about 70% at 8 hours, between about 25% and about 80% at 12 hours, between about 35% to about 90% at 16 hours, between about 55% and about 100% hours at 24 hours and greater than 85% at 36 hours; or between 0% and about 40% at 1 hour, between about 0% and about 55% at 2 hours, between about 10% and about 65% at 4 hours, between about 20% and about 75% at 8 hours, between about 30% and about 85% at 12 hours, between about 40% to about 100% at 16 hours, between about 55% and about 100% hours at 24 hours and greater than 90% at 36 hours; or between 0% and about 45% at 1 hour, between about 0% and about 60% at 2 hours, between about 15% and about 70% at 4 hours, between about 25% and about 80% at 8 hours, between about 35% and about 90% at 12 hours, between about 45% to about 100% at 16 hours, between about 60% and about 100% hours at 24 hours and greater than 60% at 36 hours; or between 0% and about 50% at 1 hour, between about 5% and about 65% at 2 hours, between about 20% and about 75% at 4 hours, between about 30% and about 85% at 8 hours, between about 40% and about 95% at 12 hours, between about 50% to about 100% at 16 hours, between about 70% and about 100% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 30% at 1 hour, between about 5% and about 40% at 2 hours, between about
10% and about 60% at 4 hours, between about 20% and about 70% at 8 hours, between about 30% and about 100% at 12 hours, between about 40% to about 100% at 16 hours, between about 60% and about 100% hours at 24 hours and greater than 90% at 36 hours; or between 0% and about 30% at 1 hour, between about 0% and about 30% at 2 hours, between about 0% and about 30% at 4 hours, between about 5% and about 70% at 8 hours, between about 10% and about 80% at 12 hours, between about 20% to about 100% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 50% at 36 hours; or between 0% and about 20% at 1 hour, between about 0% and about 20% at 2 hours, between about 0% and about 20% at 4 hours, between about 0% and about 20% at 8 hours, between about 5% and about 40% at 12 hours, between about 10% to about 80% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 60% at 36 hours; or between 0% and about 10% at 1 hour, between about 0% and about 20% at 2 hours, between about 0% and about 40% at 4 hours, between about 5% and about 60% at 8 hours, between about 10% and about 80% at 12 hours, between about 20% to about 100% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 50% at 36 hours. ] In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 10% and about 100% at 8 hours, between about 20% and about 100% at 12 hours, between about 30% to about 100% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours. In other preferred embodiments, said release rate is between 0% and about 45% at 1 hour, between about 0% and about 70% at 2 hours, between about 3% and about 90% at 4 hours, between about 8% and about 100% at 8 hours, between
about 15% and about 100% at 12 hours, between about 25% to about 100% at 16 hours, between about 45% and about 100% hours at 24 hours and greater than 80% at 36 hours; or between 0% and about 40% at 1 hour, between about 0% and about 65% at 2 hours, between about 0% and about 80% at 4 hours, between about 5% and about 80% at 8 hours, between about 10% and about 90% at 12 hours, between about 20% to about 100% at 16 hours, between about 40% and about 100% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 35% at 1 hour, between about 0% and about 60% at 2 hours, between about 0% and about 70% at 4 hours, between about 3% and about 70% at 8 hours, between about 5% and about 80% at 12 hours, between about 15% to about 100% at 16 hours, between about 30% and about 100% hours at 24 hours and greater than 40% at 36 hours; or between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 5% and about 100% at 4 hours, between about 15% and about 100% at 8 hours, between about 30% and about 100% at 12 hours, between about 40% to about 100% at 16 hours, between about 60% and about 100% hours at 24 hours and greater than 70% at 36 hours; or between 0% and about 50% at 1 hour, between about 0% and about 75% at 2 hours, between about 5% and about 95% at 4 hours, between about 25% and about 80% at 8 hours, between about 30% and about 100% at 12 hours, between about 40% to about 100% at 16 hours, between about 60% and about 100% hours at 24 hours and greater than 60% at 36 hours; or between 0% and about 60% at 1 hour, between about 0% and about 85% at 2 hours, between about 5% and about 100% at 4 hours, between about 10% and about 100% at 8 hours, between about 20% and about 100% at 12 hours, between about 30% to about 100% at 16 hours, between about 50% and about 100% hours at 24 hours and greater than 80% at 36 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said opioid agonist dosage form providing an in-vitro release rate by weight of the opioid agonist, when measured by the USP Basket or Paddle
Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 15% and about 25% at 1 hour, between about 25% and about 35% at 2 hours, between about 30% and about 45% at 4 hours, between about 40% and about 60% at 8 hours, between about 55% and about 70% at 12 hours and between about 60% to about 75% at 16 hours. In other preferred embodiments, said release rate is between 10% and about 20% at 1 hour, between about 20% and about 30% at 2 hours, between about 25% and about 40% at 4 hours, between about 30% and about 50% at 8 hours, between about 50% and about 65% at 12 hours and between about 55% to about 65% at 16 hours; or between 5% and about 15% at 1 hour, between about 15% and about 25% at 2 hours, between about 20% and about 35% at 4 hours, between about 25% and about 45% at 8 hours, between about 45% and about 60% at 12 hours and between about 50% to about 60% at 16 hours; or between 15% and about 30% at 1 hour, between about 20% and about 40% at 2 hours, between about 20% and about 50% at 4 hours, between about 30% and about 70% at 8 hours, between about 60% and about 80% at 12 hours and between about 70% to about 90% at 16 hours; or between 0% and about 50% at 1 hour, between about 5% and about 50% at 2 hours, between about 5% and about 70% at 4 hours, between about 10% and about 80% at 8 hours, between about 20% and about 100% at 12 hours and between about 40% to about 100% at 16 hours; or between 15% and about 40% at 1 hour, between about 15% and about 45% at 2 hours, between about 20% and about 60% at 4 hours, between about 20% and about 80% at 8 hours, between about 30% and about 90% at 12 hours and between about 40% to about 100% at 16 hours. In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said in-vitro release rate being substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than
30%. In other preferred embodiments, the difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH using the aforementioned methods is no greater than 50%, or no greater than 4,0%, or no greater than 35%, or no greater than 25%, or no greater than 20%, or no greater than 15%, or no greater than 10%, or no greater than 5%.
[00379] In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said dosage forms of opioid agonist providing in-vitro release rates by weight of between 0% to about 50% by weight of the opioid agonist from the dosage form at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C. In other preferred embodiments, said release rate at one hour is between 0% to about 10% by weight, or 0% to about 20% by weight, or is between 0% to about 30% by weight, or 0% to about 40% by weight, or between 0% to about 60% by weight, or 0% to about 70% by weight, or 0% to about 80% by weight, or 0% to about 90% by weight, or 10% to about 50% by weight, or 10% to about 60% by weight, or 10% to about 70% by weight, or 10% to about 90% by weight, or 10% to about 100% by weight, or 30% to about 100% by weight, or 50% to about 100% by weight.
[00380] In some preferred embodiments, the dosage form provides a pharmaceutical composition comprising a therapeutically effective amount of opioid agonist or pharmaceutically acceptable salts thereof or mixtures thereof, said dosage forms of opioid agonist providing in-vitro release rates by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of between 0% to about 80% at 0.5 hours, and greater than about 40% at 1 hour. In other preferred embodiments, said release rate is between 0% to about 40% at 0.5 hours, and greater than about 60% at 1 hour; or between 0% to about 20% at 0.5 hours, and greater than about 40% at 1 hour; or between 0% to about 20% at 0.5 hours, and greater than about 20% at 1 hour; or
between 0% to about 90% at 0.5 hours, and greater than about 60% at 1 hour; or between 0% to about 100% at 0.5 hours, and greater than about 60% at 1 hour; or between 0% to about 90% at 1 hour, and greater than about 40% at 2 hours; or between 0% to about 100% at 1 hour, and greater than about 60% at 2 hours; or between 0% to about 60% at 1 hour, and greater than about 40% at 2 hours; or between 0% to about 40% at 1 hour, and greater than about 30% at 2 hours; or between 0% to about 50% at 1 hour, and greater than about 40% at 2 hours; or between 0% to about 30% at 1 hour, and greater than about 20% at 2 hours; or between 0% and about 50% at 1 hour, between about 0% and about 80% at 2 hours, between about 5% and about 100% at 4 hours and between about 10% and about 100% at 8 hours; or between 10% and about 60% at 1 hour, between about 15% and about 75% at 2 hours, between about 20% and about 95% at 4 hours and between about 30% and about 100% at 8 hours.
[00381] In some preferred embodiments, some or all of the dissolution embodiments and specifications (e.g., USP Basket Method, USP Paddle Method) of the invention applicable to the opioid agonist of the oral dosage are also applicable to the substantially non-releasable or non-releasable aversive agent (e.g., cannabinoid antagonist or alcohol deterrent) of the dosage form. In the interest of brevity, said aversive agent dissolution embodiments and specifications are not repeated.
[00382] In some preferred embodiments, some or all of the pharmacokinetic embodiments and specifications (e.g., Cmax, Tmax, AUC, percent absorption) of the invention applicable to the opioid agonist of the oral dosage are also applicable to the substantially non-releasable or non-releasable aversive agent (e.g., cannabinoid antagonist or alcohol deterrent) of the dosage form. In the interest of brevity, said aversive agent dissolution embodiments and specifications are not repeated.
Alkylcellulose Polymers
[00383] Cellulosic materials and polymers, including alkylcelluloses, provide hydrophobic materials well suited for coating the beads according to the invention. Simply by way of example, one preferred alkylcellulosic polymer is
ethylcellulose, although the artisan will appreciate that other cellulose and/or alkylcellulose polymers may be readily employed, singly or in any combination, as all or part of a hydrophobic coating according to the invention.
[00384] One commercially-available aqueous dispersion of ethylcellulose is
Aquacoat. Aquacoat is prepared by dissolving the ethylcellulose in a water- immiscible organic solvent and then emulsifying the same in water in the presence of a surfactant and a stabilizer. After homogenization to generate submicron droplets, the organic solvent is evaporated under vacuum to form a pseudolatex. The plasticizer is not incorporated in the pseudolatex during the manufacturing phase. Thus, prior to using the same as a coating, it is necessary to intimately mix the Aquacoat with a suitable plasticizer prior to use.
[00385] Another aqueous dispersion of ethylcellulose is commercially available as Surelease. This product is prepared by incorporating plasticizer into the dispersion during the manufacturing process. A hot melt of a polymer, plasticizer (dibutyl sebacate), and stabilizer (oleic acid) is prepared as a homogeneous mixture, which is then diluted with an alkaline solution to obtain an aqueous dispersion which can be applied directly onto substrates.
Acrylic Polymers
[00386] In other preferred embodiments of the present invention, the hydrophobic material comprising the controlled release coating is a pharmaceutically acceptable acrylic polymer, including but not limited to acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamide copolymer, poly(methyl methacrylate), polymethacrylate, poly(methyl methacrylate) copolymer, polyacrylamide, aminoalkyl methacrylate copolymer, poly(methacrylic acid anhydride), and glycidyl methacrylate copolymers.
[00387] In certain preferred embodiments, the acrylic polymer is comprised of one or more ammonio methacrylate copolymers. Ammonio methacrylate copolymers are well known in the art, and are described in NF XVII as fully
polymerized copolymers of acrylic and methacrylic acid esters with a low content of quaternary ammonium groups.
[00388] In order to obtain a desirable dissolution profile, it may be necessary to incorporate two or more ammonio methacrylate copolymers having differing physical properties, such as different molar ratios of the quaternary ammonium groups to the neutral (meth)acrylic esters.
[00389] Certain methacrylic acid ester-type polymers are useful for preparing pH-dependent coatings which may be used in accordance with the present invention. For example, there are a family of copolymers synthesized from diethylaminoethyl methacrylate and other neutral methacrylic esters, also known as methacrylic acid copolymer or polymeric methacrylates, commercially available as Eudragit. There are several different types of Eudragit. For example, Eudragit E is an example of a methacrylic acid copolymer which swells and dissolves in acidic media. Eudragit L is a methacrylic acid copolymer which does not swell at about pH<5.7 and is soluble at about pH>6. Eudragit S does not swell at about pH<6.5 and is soluble at about pH>7. Eudragit RL and Eudragit RS are water swellable, and the amount of water absorbed by these polymers is pH-dependent, however, dosage forms coated with Eudragit RL and RS are pH-independent.
[00390] In certain preferred embodiments, the acrylic coating comprises a mixture of two acrylic resin lacquers commercially available from under the trade names Eudragit RL30D and Eudragit RS30D, respectively. Eudragit RL30D and Eudragit RS30D are copolymers of acrylic and methacrylic esters with a low content of quaternary ammonium groups, the molar ratio of ammonium groups to the remaining neutral (meth)acrylic esters being 1:20 in Eudragit RL30D and 1 :40 in Eudragit RS30D. The mean molecular weight is about 150,000. The code designations RL (high permeability) and RS (low permeability) refer to the permeability properties of these agents. Eudragit RL/RS mixtures are insoluble in water and in digestive fluids. However, coatings formed from the same are swellable and permeable in aqueous solutions and digestive fluids.
[00391] The Eudragit RL/RS dispersions of the present invention may be mixed together in any desired ratio in order to ultimately obtain a sustained release formulation having a desirable dissolution profile. Desirable sustained release formulations may be obtained, for instance, from a retardant coating derived from 100% Eudragit RL, 50% Eudragit RL and 50% Eudragit RS, and 10% Eudragit RL:90%Eudragit RS. Of course, one skilled in the art will recognize that other acrylic polymers may also be used, such as, for example, Eudragit L.
Plasticizers
[00392] In embodiments of the present invention where the coating comprises an aqueous dispersion of a hydrophobic material, the inclusion of an effective amount of a plasticizer in the aqueous dispersion of hydrophobic material will further improve the physical properties of the sustained release coating. For example, because ethylcellulose has a relatively high glass transition temperature and does not form flexible films under normal coating conditions, it is preferable to incorporate a plasticizer into an ethylcellulose coating containing sustained release coating before using the same as a coating material. Generally, the amount of plasticizer included in a coating solution is based on the concentration of the film-former, e.g., most often from about 1 to about 50 percent by weight of the film-former. Concentration of the plasticizer, however, can only be properly determined after careful experimentation with the particular coating solution and method of application.
[00393] Examples of suitable plasticizers for ethylcellulose include water insoluble plasticizers such as dibutyl sebacate, diethyl phthalate, triethyl citrate, tributyl citrate, and triacetin, although it is possible that other water- insoluble plasticizers (such as acetylated monoglycerides, phthalate esters, castor oil, etc.) may be used. Triethyl citrate is an especially preferred plasticizer for the aqueous dispersions of ethyl cellulose of the present invention.
[00394] Examples of suitable plasticizers for the acrylic polymers of the present invention include, but are not limited to citric acid esters such as triethyl citrate NF XVI, tributyl citrate, dibutyl phthalate, and possibly 1,2- propylene glycol. Other plasticizers which have proved to be suitable for enhancing the elasticity of the films formed from acrylic films such as Eudragit RL/RS lacquer solutions include polyethylene glycols, propylene glycol, diethyl phthalate, castor oil, and triacetin. Triethyl citrate is an especially preferred plasticizer for the aqueous dispersions of ethyl cellulose of the present invention.
[00395] It has further been found that the addition of a small amount of talc reduces the tendency of the aqueous dispersion to stick during processing, and acts as a polishing agent.
Processes for Preparing Coated Beads
[00396] When a hydrophobic controlled release coating material is used to coat inert pharmaceutical beads such as nu pariel 18/20 beads, which are already coated with an opioid agonist, a plurality of the resultant solid controlled release beads may thereafter be placed in a gelatin capsule, with the cannabinoid antagonist in a substantially non-releasable form. The dosage form provides an effective controlled release dose of the opioid agonist when ingested and contacted by an environmental fluid, e.g., gastric fluid or dissolution media.
[00397] The controlled release bead formulations of the present invention slowly release the opioid agonist, e.g., when ingested and exposed to gastric fluids, and then to intestinal fluids. The controlled release profile of the formulations of the invention can be altered, for example, by varying the amount of overcoating with the hydrophobic material, altering the manner in which the plasticizer is added to the hydrophobic material, by varying the amount of plasticizer relative to hydrophobic material, by the inclusion of additional ingredients or excipients, by altering the method of manufacture, etc. The dissolution profile of the ultimate product may also be modified, for example, by increasing or decreasing the thickness of the retardant coating.
[00398) Spheroids or beads coated with an opioid agonist may be prepared, e.g., by dissolving the drug in water and then spraying the solution onto a substrate, for example, nu pariel 18/20 beads, using a Wuster insert. Optionally, additional ingredients are also added prior to coating the beads in order to assist the binding of the cannabinoid to the beads, and/or to color the solution, etc. For example, a product which includes hydroxypropyl methylcellulose, etc. with or without colorant (e.g., Opadry) may be added to the solution and the solution mixed (e.g., for about 1 hour) prior to application of the same onto the beads. The resultant coated substrate, in this example beads, may then be optionally overcoated with a barrier agent, to separate the therapeutically active agent from the hydrophobic controlled release coating. An example of a suitable barrier agent is one which comprises hydroxypropyl methylcellulose. However, any film-former known in the art may be used. It is preferred that the barrier agent does not affect the dissolution rate of the final product.
[00399] The beads may then be overcoated with an aqueous dispersion of the hydrophobic material. The aqueous dispersion of hydrophobic material preferably further includes an effective amount of plasticizer, e.g. triethyl citrate. Pre-formulated aqueous dispersions of ethylcellulose, such as Aquacoat or Surelease, may be used. If Surelease is used, it is not necessary to separately add a plasticizer. Alternatively, pre-formulated aqueous dispersions of acrylic polymers such as Eudragit can be used.
[004001 The coating solutions of the present invention preferably contain, in addition to the film-former, plasticizer, and solvent system (i.e., water), a colorant to provide elegance and product distinction. Color may be added to the solution of the therapeutically active agent instead, or in addition to the aqueous dispersion of hydrophobic material. For example, color may be added to Aquacoat via the use of alcohol or propylene glycol based color dispersions, milled aluminum lakes and opacifϊers such as titanium dioxide by adding color with shear to water soluble polymer solution and then using low shear to the plasticized Aquacoat. Alternatively, any suitable method of providing color to the formulations of the present invention may be used. Suitable ingredients for
providing color to the formulation when an aqueous dispersion of an acrylic polymer is used include titanium dioxide and color pigments, such as iron oxide pigments. The incorporation of pigments, may, however, increase the retard effect of the coating.
[00401] Plasticized hydrophobic material may be applied onto the substrate comprising the therapeutically active agent by spraying using any suitable spray equipment known in the art. In a preferred method, a Wurster fluidized- bed system is used in which an air jet, injected from underneath, fluidizes the core material and effects drying while the acrylic polymer coating is sprayed on. A sufficient amount of the hydrophobic material to obtain a predetermined controlled release of said therapeutically active agent when the coated substrate is exposed to aqueous solutions, e.g. gastric fluid, is preferably applied, taking into account the physical characteristics of the therapeutically active agent, the manner of incorporation of the plasticizer, etc. After coating with the hydrophobic material, a further overcoat of a film-former, such as Opadry, is optionally applied to the beads. This overcoat is provided, if at all, in order to substantially reduce agglomeration of the beads.
[00402] The release of the therapeutically active agent from the controlled release formulation of the present invention can be further influenced, i.e., adjusted to a desired rate, by the addition of one or more release-modifying agents, or by providing one or more passageways through the coating. The ratio of hydrophobic material to water soluble material is determined by, among other factors, the release rate required and the solubility characteristics of the materials selected.
[00403] The release-modifying agents which function as pore-formers may be organic or inorganic, and include materials that can be dissolved, extracted or leached from the coating in the environment of use. The pore-formers may comprise one or more hydrophilic materials such as hydroxypropyl methylcellulose.
[00404] The sustained release coatings of the present invention can also include erosion-promoting agents such as starch and gums.
[00405] The sustained release coatings of the present invention can also include materials useful for making microporous lamina in the environment of use, such as polycarbonates comprised of linear polyesters of carbonic acid in which carbonate groups reoccur in the polymer chain. The release-modifying agent may also comprise a semi-permeable polymer. In certain preferred embodiments, the release-modifying agent is selected from hydroxypropyl methylcellulose, lactose, metal stearates, and mixtures of any of the foregoing.
[00406] The sustained release coatings of the present invention may also include an exit means comprising at least one passageway, orifice, or the like. The passageway may be formed by such methods as those disclosed in U.S. Pat. Nos. 4,063,064 and 4,088,864 (all of which are hereby incorporated by reference). The passageway can have any shape such as round, triangular, square, elliptical, irregular, etc.
Matrix Formulations
[00407] In other embodiments of the present invention, the controlled release formulation is achieved via a matrix having a controlled release coating as set forth above. The present invention also comprises sustained-release tablets comprising an opioid agonist and cannabinoid antagonist particles coated with a coating that renders the antagonist substantially non-releasable, wherein the agonist and the antagonist are dispersed in a controlled release matrix that affords in-vitro dissolution rates of the opioid agonist within the preferred ranges and that releases the opioid agonist in a pH-dependent or pH- independent manner. The materials suitable for inclusion in a controlled release matrix will depend on the method used to form the matrix.
[00408] For example, a matrix in addition to the opioid agonist and the substantially non-releasable form of the coated cannabinoid antagonist, may include: (i) Hydrophilic and/or hydrophobic materials, such as gums, cellulose ethers, acrylic resins, protein derived materials; the list is not meant to be exclusive, and any pharmaceutically acceptable hydrophobic material or hydrophilic material which is capable of imparting controlled release of the cannabinoid may be used in accordance with the present invention; (2)
Digestible, long chain (C8 -C50, especially Ci2-C40), substituted or unsubstituted hydrocarbons, such as fatty acids, fatty alcohols, glyceryl esters of fatty acids, mineral and vegetable oils and waxes, and stearyl alcohol; and polyalkylene glycols.
[00409] Of these polymers, especially Eudragit RSPO, the cellulose ethers, especially hydroxyalkyl celluloses and carboxyalkyl celluloses, are preferred. The oral dosage form may contain between 1% and 80% (by weight) of at least one hydrophilic or hydrophobic material.
[00410] When the hydrophobic material is a hydrocarbon, the hydrocarbon preferably has a melting point of between 25°C and 90°C. Of the long chain hydrocarbon materials, fatty (aliphatic) alcohols are preferred. The oral dosage form may contain up to 60% (by weight) of at least one digestible, long chain hydrocarbon.
[00411] Preferably, the oral dosage form contains up to 60% (by weight) of at least one polyalkylene glycol.
[00412] The hydrophobic material is preferably selected from the group consisting of alkylcelluloses, acrylic and methacrylic acid polymers and copolymers, shellac, zein, hydrogenated castor oil, hydrogenated vegetable oil, or mixtures thereof. In certain preferred embodiments of the present invention, the hydrophobic material is a pharmaceutically acceptable acrylic polymer, including but not limited to acrylic acid and methacrylic acid copolymers, methyl methacrylate, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, aminoalkyl methacrylate copolymer, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamine copolymer, poly(methyl methacrylate), poly(methacrylic acid)(anhydride), polymethacrylate, polyacrylamide, poly(methacrylic acid anhydride), and glycidyl methacrylate copolymers. In other embodiments, the hydrophobic material is selected from materials such as hydroxyalkylcelluloses such as hydroxypropylmethylcellulose and mixtures of the foregoing.
[00413] Preferred hydrophobic materials are water-insoluble with more or less pronounced hydrophilic and/or hydrophobic trends. Preferably, the hydrophobic materials useful in the invention have a melting point from about
3O0C to about 2000C, preferably from about 45°C to about 900C. Specifically, the hydrophobic material may comprise natural or synthetic waxes, fatty alcohols (such as lauryl, myristyl, stearyl, cetyl or preferably cetostearyl alcohol), fatty acids, including but not limited to fatty acid esters, fatty acid glycerides (mono-, di-, and ϋϊ-glycerides), hydrogenated fats, hydrocarbons, normal waxes, stearic aid, stearyl alcohol and hydrophobic and hydrophilic materials having hydrocarbon backbones. Suitable waxes include, for example, beeswax, glycowax, castor wax and camauba wax. For purposes of the present invention, a wax-like substance is defined as any material which is normally solid at room temperature and has a melting point of from about 3O0C to about 1000C.
[00414] Suitable hydrophobic materials which may be used in accordance with the present invention include digestible, long chain (C8-C5O, especially Ci2- C40), substituted or unsubstituted hydrocarbons, such as fatty acids, fatty alcohols, glyceryl esters of fatty acids, mineral and vegetable oils and natural and synthetic waxes. Hydrocarbons having a melting point of between 250C and 9O0C are preferred. Of the long chain hydrocarbon materials, fatty (aliphatic) alcohols are preferred in certain embodiments. The oral dosage form may contain up to 60% (by weight) of at least one digestible, long chain hydrocarbon.
(00415] Preferably, a combination of two or more hydrophobic materials are included in the matrix formulations. If an additional hydrophobic material is included, it is preferably selected from natural and synthetic waxes, fatty acids, fatty alcohols, and mixtures of the same. Examples include beeswax, camauba wax, stearic acid and stearyl alcohol. This list is not meant to be exclusive.
[00416] One particular suitable matrix comprises at least one water soluble hydroxyalkyl cellulose, at least one Ci2-C36, preferably CH-C22, aliphatic alcohol and, optionally, at least one polyalkylene glycol. The at least one hydroxyalkyl cellulose is preferably a hydroxy (Ci to C6) alkyl cellulose, such as hydroxypropylcellulose, hydroxypropylmethylcellulose and, especially, hydroxyethylcellulose. The amount of the at least one hydroxyalkyl cellulose
in the present oral dosage form will be determined, inter alia, by the precise rate of cannabinoid release required. The at least one aliphatic alcohol may be, for example, lauryl alcohol, myristyl alcohol or stearyl alcohol. In particularly preferred embodiments of the present oral dosage form, however, the at least one aliphatic alcohol is cetyl alcohol or cetostearyl alcohol. The amount of the at least one aliphatic alcohol in the present oral dosage form will be determined, as above, by the precise rate of cannabinoid release required. It will also depend on whether at least one polyalkylene glycol is present in or absent from the oral dosage form. In the absence of at least one polyalkylene glycol, the oral dosage form preferably contains between 20% and 50% (by wt) of the at least one aliphatic alcohol. When at least one polyalkylene glycol is present in the oral dosage form, then the combined weight of the at least one aliphatic alcohol and the at least one polyalkylene glycol preferably constitutes between 20% and 50% (by wt) of the total dosage.
[00417] In one embodiment, the ratio of, e.g., the at least one hydroxyalkyl cellulose or acrylic resin to the at least one aliphatic alcohol/polyalkylene glycol determines, to a considerable extent, the release rate of the cannabinoid from the formulation. A ratio of the at least one hydroxyalkyl cellulose to the at least one aliphatic alcohol/polyalkylene glycol of between 1 :2 and 1 :4 is preferred, with a ratio of between 1 :3 and 1 :4 being particularly preferred.
[00418] The at least one polyalkylene glycol may be, for example, polypropylene glycol or, which is preferred, polyethylene glycol. The number average molecular weight of the at least one polyalkylene glycol is preferred between 1,000 and 15,000 especially between 1,500 and 12,000.
[00419] Another suitable controlled release matrix would comprise an alkylcellulose (especially ethyl cellulose), a Ci2 to C36 aliphatic alcohol and, optionally, a polyalkylene glycol.
[00420] In another preferred embodiment, the matrix includes a pharmaceutically acceptable combination of at least two hydrophobic materials.
[00421] In addition to the above ingredients, a controlled release matrix may also contain suitable quantities of other materials, e.g. diluents, lubricants,
binders, granulating aids, colorants, flavorants and glidants that are conventional in the pharmaceutical art.
Processes for Preparing Matrix— Based Beads
[00422] In order to facilitate the preparation of a solid, controlled release, oral dosage form according to this invention, any method of preparing a matrix formulation known to those skilled in the art may be used. For example incorporation in the matrix may be effected, for example, by (a) forming granules comprising at least one water soluble hydroxyalkyl cellulose and cannabinoid or a cannabinoid salt; (b) mixing the hydroxyalkyl cellulose containing granules with at least one Ci2-C36 aliphatic alcohol; and (c) optionally, compressing and shaping the granules. Preferably, the granules are formed by wet granulating the hydroxy-alkyl cellulose/cannabinoid with water. In a particularly preferred embodiment of this process, the amount of water added during the wet granulation step is preferably between 1.5 and 5 times, especially between 1.7"5 and 3.5 times, the dry weight of the cannabinoid.
[00423] In yet other alternative embodiments, a spheronizing agent, together with the active ingredient can be spheronized to form spheroids. Macrocrystalline cellulose is preferred. A suitable microcrystalline cellulose is, for example, the material sold as Avicel PH 101. In such embodiments, in addition to the active ingredient and- spheronizing agent, the spheroids may also contain a binder. Suitable binders, such as low viscosity, water soluble polymers, will be well known to those skilled in the pharmaceutical art. However, water soluble hydroxy lower alkyl cellulose, such as hydroxypropylcellulose, are preferred. Additionally (or alternatively) the spheroids may contain a water insoluble polymer, especially an acrylic polymer, an acrylic copolymer, such as a methacrylic acid-ethyl acrylate copolymer, or ethyl cellulose. In such embodiments, the sustained release coating will generally include a hydrophobic material such as (a) a wax, either alone or in admixture with a fatty alcohol; or (b) shellac or zein.
Melt Extrusion Matrix
[00424J Sustained release matrices can also be prepared via melt-granulation or melt-extrusion techniques, as long as the techniques used do not damage the integrity of the substantially non-releasable form of the cannabinoid antagonist added during the preparation of the matrix to the extent that sufficient amount of the cannabinoid antagonist becomes available to be released into the gastrointestinal system upon oral administration. Alternatively, the melt extrusion step may be performed with the opioid agonist to produce sustained release particles of the agonist, which may then be combined with the substantially non-releasable form of the cannabinoid antagonist. Generally, melt-granulation techniques involve melting a normally solid hydrophobic material, e.g. a wax, and incorporating a powdered drug therein. To obtain a sustained release dosage form, it may be necessary to incorporate an additional hydrophobic substance, e.g. ethylcellulose or a water-insoluble acrylic polymer, into the molten wax hydrophobic material. Examples of sustained release formulations prepared via melt-granulation techniques are found in U.S. Pat. No. 4,861,598, and hereby incorporated by reference in its entirety.
(00425] The additional hydrophobic material may comprise one or more water- insoluble wax-like thermoplastic substances possibly mixed with one or more wax-like thermoplastic substances being less hydrophobic than said one or more water-insoluble wax-like substances. In order to achieve constant release, the individual wax-like substances in the formulation should be substantially non-degradable and insoluble in gastrointestinal fluids during the initial release phases. Useful water-insoluble wax-like substances may be those with a water-solubility that is lower than about 1 : 5,000 (w/w).
[00426] In addition to the above ingredients, a sustained release matrix may also contain suitable quantities of other materials, e.g., diluents, lubricants, binders, granulating aids, colorants, flavorants and glidants that are conventional in the pharmaceutical art. The quantities of these additional materials will be sufficient to provide the desired effect to the desired formulation.
[00427] In addition to the above ingredients, a sustained release matrix incorporating melt-extruded multiparticulates may also contain suitable quantities of other materials, e.g. diluents, lubricants, binders, granulating aids, colorants, flavorants and glidants that are conventional in the pharmaceutical art in amounts up to about 50% by weight of the particulate if desired.
[00428] Specific examples of pharmaceutically acceptable carriers and excipients that may be used to formulate oral dosage forms are described in the Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), incorporated by reference herein.
Melt Extrusion Multiparticulates
[00429] The preparation of a suitable melt-extruded matrix according to the present invention may, for example, include the steps of blending the cannabinoid analgesic, together with at least one hydrophobic material and preferably the additional hydrophobic material to obtain a homogeneous mixture. The homogeneous mixture is then heated to a temperature sufficient to at least soften the mixture sufficiently to extrude the same. The resulting homogeneous mixture is then extruded to form strands. The extrudate is preferably cooled and cut into multiparticulates by any means known in the art. The strands are cooled and cut into multiparticulates. The multiparticulates are then blended with the cannabinoid antagonist particles coated with a coating that renders the antagonist substantially non-releasable and divided into unit doses. The extrudate preferably has a diameter of from about 0.1 to about 5 mm and provides sustained release of the opioid agonist for a time period of from about 8 to about 24 hours.
[00430] An optional process for preparing the melt extrusions of the present invention includes directly metering into an extruder a hydrophobic material, a therapeutically active agent, and an optional binder; heating the homogenous mixture; extruding the homogenous mixture to thereby form strands; cooling the strands containing the homogeneous mixture; cutting the strands into particles having a size from about 0.1 mm to about 12 mm; and combining the particles with the coated cannabinoid antagonist particles and dividing them
into unit doses. In this aspect of the invention, a relatively continuous manufacturing procedure is realized.
[00431] The diameter of the extruder aperture or exit port can also be adjusted to vary the thickness of the extruded strands. Furthermore, the exit part of the extruder need not be round; it can be oblong, rectangular, etc. The exiting strands can be reduced to particles using a hot wire cutter, guillotine, etc.
[00432] The melt extruded multiparticulate system can be, for example, in the form of granules, spheroids or pellets depending upon the extruder exit orifice. For purposes of the present invention, the terms "melt-extruded multiparticulate(s)" and "melt-extruded multiparticulate system(s)" and "melt- extruded particles" shall refer to a plurality of units, preferably within a range of similar size and/or shape and containing one or more active agents and one or more excipients, preferably including a hydrophobic material as described herein. In this regard, the melt-extruded multiparticulates will be of a range of from about 0.1 to about 12 mm in length and have a diameter of from about 0.1 to about 5 mm. In addition, it is to be understood that the melt-extruded multiparticulates can be any geometrical shape within this size range. Alternatively, the extrudate may simply be cut into desired lengths and divided into unit doses of the therapeutically active agent without the need of a spheronization step.
[00433] In one preferred embodiment, oral dosage forms are prepared to include an effective amount of melt-extruded multiparticulates within a capsule. For example^ a plurality of the melt-extruded multiparticulates may be placed in a gelatin capsule in an amount sufficient to provide an effective sustained release dose when ingested and contacted by gastric fluid.
[00434] In another preferred embodiment, a suitable amount of the multiparticulate extrudate is combined with the coated cannabinoid antagonist particles and compressed into an oral tablet using conventional tableting equipment using standard techniques. Techniques and compositions for making tablets (compressed and molded), capsules (hard and soft gelatin) and pills are also described in Remington's Pharmaceutical Sciences, incorporated by reference herein.
[00435] In yet another preferred embodiment, the coated cannabinoid antagonist particles are added during the extrusion process and the extrudate can be shaped into tablets as set forth in U.S. Pat. No. 4,957,681, described in additional detail above and hereby incorporated by reference.
[00436] Optionally, the sustained release melt-extruded multiparticulate systems or tablets can be coated, or the gelatin capsule can be further coated, with a sustained release coating such as the sustained release coatings described above. Such coatings preferably include a sufficient amount of hydrophobic material to obtain a weight gain level from about 2 to about 30 percent, although the overcoat may be greater depending upon the physical properties of the particular cannabinoid analgesic compound utilized and the desired release rate, among other things.
[00437] The melt-extruded unit dosage forms of the present invention may further include combinations of melt-extruded multiparticulates containing one or more of the therapeutically active agents disclosed above before being encapsulated. Furthermore, the unit dosage forms can also include an amount of an immediate release opioid agonist for prompt therapeutic effect. The immediate release opioid agonist may be incorporated, e.g., as separate pellets within a gelatin capsule, or may be coated on the surface of the multiparticulates after preparation of the dosage forms (e.g., controlled release coating or matrix-based). The unit dosage forms of the present invention may also contain a combination of controlled release beads and matrix multiparticulates to achieve a desired effect.
[00438] The sustained release formulations of the present invention preferably slowly release the opioid agonist, e.g., when ingested and exposed to gastric fluids, and then to intestinal fluids. The sustained release profile of the melt- extruded formulations of the invention can be altered, for example, by varying the amount of retardant, i.e., hydrophobic material, by varying the amount of plasticizer relative to hydrophobic material, by the inclusion of additional ingredients or excipients, by altering the method of manufacture, etc.
[00439] In other embodiments of the invention, the melt extruded material is prepared without the inclusion of the opioid agonist and/or coated cannabinoid
antagonist particles, which are added thereafter to the extrudate. Such formulations typically will have the drugs blended together with the extruded matrix material, and then the mixture would be tableted in order to provide a slow release of the opioid agonist. Such formulations may be advantageous, for example, when the therapeutically active agent included in the formulation is sensitive to temperatures needed for softening the hydrophobic material and/or the retardant material.These and other embodiments of the present invention will readily occur to those of ordinary skill in the art in view of the disclosure herein.
Solid Dispersions
[00440] In addition to the foregoing methods, in some embodiments, when the non-releasable aversive agent is a liquid, or a semisolid or a poorly water soluble liquid or solid, the dosage form may be prepared using other methods.
[00441] In some embodiments, the dosage form may be mixed and blended, optionally in the presence of heat, with a fine powder excipient (e.g., talc), the blended material continuously fed into a twin screw extruder, and the resultant strands collected on a conveyor. The strands are allowed to cool on the conveyor and then cut into pellets using a Pelletizer. The the pellets are screened and' the desired sieve portion collected.
[00442] In some embodiments, in the examples providede herein, the water is replaced with ethanol, hydroalcoholic solutions or organic solvents know in the art to aid in the dissolution, dispersion or mixing of the aversive agent.
[00443] In other embodiments of the invention, solid dispersions of the aversive agent and HPMC are prepared by hot-stage extrusion with a corotating twin-screw extruder. The extrudates are collected after cooling at ambient temperature on a conveyor belt. Samples are milled for 1 min with a laboratory cutting mill and sieved to the desired particle size.
Detailed Description of the Preferred Embodiments
[00444] The following examples illustrate various aspects of the present invention. They are not to be construed to limit the claims in any manner whatsoever.
[00445] A wide variety of methods known in the art for the preparation of immediate release and controlled release dosage forms may be incorporated into the invention.
[00446] Other suitable substantially non-releasable forms of aversive agents
(i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents, and mixture thereof, as defined in this invention may also be prepared by modification of the examples herein and by use of material other than those specifically disclosed herein, including those which may hereafter become known to the art to be capable of performing the necessary functions.
[00447] Other suitable modifications and adaptations of the variety of conditions and parameters normally encountered and obvious to those skilled in the art are within the spirit and scope of the invention.
[00448] More than one substantially non-releasable aversive agent selected from the group comprising cannabinoid antagonists and alcohol deterrents may be included in the dosage form either in the form of separate substantially non-releasable subunits, on combined into the same substantially non- releasable subunits. Said more than one substantially non-releasable aversive agent may comprise aversive agents from the same class of agents (e.g., more than one cannabinoid antagonist, or more than one alcohol deterrent) or from a different class of agents (e.g., one cannabinoid antagonist, one alcohol deterrent) or mixtures thereof.
[00449] The percent loading of the opioid agonist agent, the substantially non- releasable aversive agent and the abuse intervention agent onto the beads may be varied depending on the physiochemical and pharmaceutical properties of said agent and ingredients (excipients), the pharmacologic effects of said agent and the desired degree of release or non-release from the dosage form.
[00450] When the aversive agent is in the form of multiparticulates, including beads and granulation individually coated with a sequestering material, it may be directly or indirectly be overcoated with the opioid agonist such that each multiparticulate contains a aversive agent and an opioid agonist and filled uncompressed into a capsule or compressed into a capsule or tablet.
[00451] The ingredients used for the preparation of the releasable opioid agonist in immediate release form or controlled release form, and the substantially non-releasable aversive agent may be modified depending on the selection, dose and desired duration of effect of the opioid agonist and the aversive agent. In some embodiments, a change in the dose or amount opioid agonist and/or aversive agent does not require a change in amount of other ingredients. In other embodiments, a proportional change in the amount of other ingredients is required to maintain the desired properties. In yet other embodiments, a change in the dose or amount opioid agonist and/or aversive agent necessitates a change in the nature and/or amount of ingredients to provide the required characteristics of the opioid agonist (e.g., immediate release, sustained release, duration of effect, rate and extent of absorption, therapeutic concentrations and effect, etc.) and aversive agent (e.g., extent of non-release or sequestration, degree of abuse deterrence, etc).
[00452] Abuse intervention agents may optionally be incorporated into the same sub-unit as the substantially non-releasable aversive agent or into a different sub-unit or into the granulation or matrix material containing the opioid agonist.
[00453] The ingredients used for the preparation of the releasable opioid agonist in immediate release form or controlled release form, and the substantially non-releasable aversive agent may be used or further modified for the preparation of in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form abuse intervention agents selected from the group comprising (i) laxatives; (ii) cutaneous vasodilators; (iii) headache producing agents; (iv) emetics, emetogenic and nausea producing compounds; (iv) bittering agents (v) mucosal, naso-mucosal, oro-mucosal, respiratory, tissue and gastrointestinal irritants; (vi) tissue
staining, non-tissue staining and beverage staining dyes, lakes and colorants; (vii) fecal and urine discolorants; (viii) malodorous agents; (ix) opioid antagonists; and (x) and (x) benzodiazepine antagonists (e.g., flumazenil), and mixtures thereof. Abuse intervention agents of the invention may also be prepared using other material known in the art, including those which may hereafter become known to the art to be capable of performing the necessary functions.
[00454] In Example 1 to 76, a substantially non-releasable forms of aversive agents (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents and mixtures thereof is prepared by coating or mixing said aversive agent with a material that renders it substantially non-releasable.
EXAMPLE 1
Ingredients Amount (mg/unit)
Aversive Agent Loading on Sphere
Rimonabant HCl 10 Sugar Spheres (30/35 mesh) 50 Opadry White Y-5-7068 2.5 Ethanol 42.5*
Sphere Overcoating
Opadry White Y-5-7068 3.02 Purified Water 17.1 1 *
Sequestration Overcoating
Eudragit RS30D 12.10 Triethyl Citrate 2.42 Talc 4.84 Purified Water 49.21 *
Final Overcoating
Opadry White Y-5-7068 4.12 Purified Water 23.35*
Total Weight 89.0 /
* In the final product in negligible quantities only as residual moisture.
[00455] Aversive Agent Loading on Sphere: Add Rimonabant HCl in ethanol with stirring, then add Opadry White and continue mixing until a homogenous
dispersion is produced. Apply the above dispersion onto the sugar spheres using a fluid bed coater. Sphere Overcoating: Disperse Opadry White in purified water with stirring and apply the dispersion over the sugar spheres loaded with Rimonabant HCl using a fluid bed coater. Sequestration Overcoating: Mix Eudragit RS30d, triethyl citrate, talc and purified water with stirring and apply the dispersion over the loaded and overcoated sugar spheres using a fluid bed coater (i.e., a fluid bed coating machine). Final Coating: Disperse Opadry White in purified water with stirring and apply the dispersion foregoing (sequestration overcoated product). Cure: Cure the foregoing spheres at 45 ° for approximately 48 to 72 hours.
EXAMPLE 2
[00456] Example 2 is prepared as in Example 1, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-rnethyl-lH-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 3
[00457] Example 3 is prepared as in Example 1, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 3b
Ingredients Amount (mg/unit)
Aversive Agent Pellets
Rimonabant HCI 10 Gelucire 44/14 35 HPMC 17.25
Pellet Overcoating
Opadry White Y-5-7068 3.02 Purified Water 17.11 *
Sequestration Overcoating
Eudragit RS30D 12.10 Triethyl Citrate 2.42 Talc 4.84 Purified Water 49.21 *
Final Overcoating
Opadry White Y-5-7068 4.12 Purified Water 23.35*
* In the final product in negligible quantities only as residual moisture.] Aversive Agent Pellets: The Gelucire 44/14 is dispensed into a mixer and heated until fully melted. The hydroxypropyl methyl cellulose (HPMC) is dispensed into the mixer. The mixing is continued until fully dispersed. The Rimonabant is dispensed into the same vessel. The mixture is thoroughly dispersed with a high shear mixer. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The Conveyor is set to provide extrudate diameter of 0.5 mm or 1 mm and the Pelletizer is set to provide pellets of approximately 0.5 or 1 mm length. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected. Pellet Overcoating: The Opadry White is dispersed in purified water with stirring and the dispersion applied over the pellets containing Rimonabant HCl using a fluid bed coater. Sequestration Overcoating: The Eudragit RS30d, triethyl citrate, talc and purified water are mixed with stirring and applied over the loaded and overcoated pellets using a fluid bed coater (i.e., a fluid bed coating machine). Final Coating: The Opadry White is dispersed in purified water with stirring and the dispersion is applied to the foregoing (sequestration
overcoated product). Cure: The foregoing pellets are cured at 45 ° for approximately 48 to 72 hours.
EXAMPLE 4
[00459] Example 4 is prepared as in Example 1, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-rnethylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 5
[00460] Example 5 is prepared as in Example 1, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 6
Example 6 is prepared as in Example 1, except that the HPMC is replaced with an equal amount of further Gelucire 44/14.
EXAMPLE 7
[00461] Example 7 is prepared as in Example 6, except that the Rimonabant
HCI 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the
group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-l h-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 8
[00462] Example 8 is prepared as in Example 6, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 9
[00463] Example 9 is prepared as in Example 1, except that the Gelucire 44/14 is replaced with an equal amount of Labrafil M 2130 CS.
EXAMPLE 10
[00464] Example 10 is prepared as in Example 9, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising * AM251, AM281 ([N-mθφholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)- 1 -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically
acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 1 1
[00465] Example 1 1 are prepared as in Example 9, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 12
[00466] Example 12 is prepared as in Example 9, except that the HPMC is replaced with an equal amount of further Labrafil M 2130 CS.
EXAMPLE 13
[00467] Example 13 is prepared as in Example 12, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 14
[00468] Example 14 is prepared as in Example 12, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group
comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 15
[00469] Example 15 is prepared as in Example 1, except that the Gelucire
44/14 is replaced with an equal amount of Beeswax.
EXAMPLE 16
[00470] Example 16 is prepared as in Example 15, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 17
[00471] Example 17 is prepared as in Example 15, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 18
[00472] Example 18 is prepared as in Example 15, except that the HPMC is replaced with an equal amount of further Beeswax.
EXAMPLE 19
[00473] Example 19 is prepared as in Example 18, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-3-hexyl-lh-l ,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 20
[00474] Example 20 is prepared as in Example 18, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 21
[00475] Example 21 is prepared as in Example 1, except that the Gelucire
44/14 is replaced with an equal amount of Cithrol GMS.
EXAMPLE 22
[00476] Example 22 is prepared as in Example 21, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SRl 44528
([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 23
[00477] Example 23 is prepared as in Example 21, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 24
[00478] Example 24 is prepared as in Example 1, except that the HPMC is replaced with an equal amount of further Cithrol GMS.
EXAMPLE 25
[00479] Example 25 is prepared as in Example 24, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxarnide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SRl 41716, SR 144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes,
polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 26
[00480] Example 26 is prepared as in Example 24, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 27
[00481] Example 27 is prepared as in Example 1, except that the Gelucire
44/14 is replaced with an equal amount of Hydrokote 1 12.
EXAMPLE 28
[00482] Example 28 is prepared as in Example 27, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SRl 44528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR 144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-3-hexyl-lh-l ,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 29
[00483] Example 30 is prepared as in Example 27, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically
acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 30
[00484] Example 30 is prepared as in Example 27, except that the HPMC is replaced with an equal amount of further Hydrokote 1 12.
EXAMPLE 31
[00485] Example 31 is prepared as in Example 27, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 32
[00486] Example 32 is prepared as in Example 27, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 33
[00487] Example 33 is prepared as in Example 1, except that the Gelucire
44/14 is replaced with an equal amount of glyceryl behenate (Compitrol™ 888 ATO).
EXAMPLE 34
[00488] Example 34 is prepared as in Example 33, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-rnethyl-lH-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methy 1-pheny I)- 1 -(4-methy lbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 35
[00489] Example 35 is prepared as in Example 33, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 36
[00490] Example 36 is prepared as in Example 33, except that the HPMC is replaced with an equal amount of further glyceryl behenate (Compitrol™ 888 ATO).
EXAMPLE 37
[00491] Example 37 is prepared as in Example 33, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-moφholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528
([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, SR141716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 38
[00492] Example 38 is prepared as in Example 33, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 39
[00493] Example 39 is prepared as in Example 1, except that the Gelucire
44/14 is replaced with an equal amount of glyceryl palmitostearate (Precirol™ ATO 5).
EXAMPLE 40
[00494] Example 40 is prepared as in Example 39, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[( 1 S)-endo- 1 ,3,3-trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l -(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR 144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes,
polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 41
[00495] Example 41 is prepared as in Example 39, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 42
[00496] Example 42 is prepared as in Example 39, except that the HPMC is replaced with an equal amount of further glyceryl palmitostearate (Precirol™ ATO 5).
EXAMPLE 43
[00497] Example 43 is prepared as in Example 39, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SRl 44528 ([N-^l S^endo-l^^-trimethylbicyc^^.^heptan^-y^^-chloro-S- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 44
[00498] Example 44 is prepared as in Example 39, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group
comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 45
[00499] Example 45 is prepared as in any of Examples 6, 12, 18, 24, 30, 36 and
42, except that the HPMC is replaced with 4 mg of 2-vinylpyrrolidone.
EXAMPLE 46
[00500] Example 46 is prepared as in Example 45, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methy 1-pheny I)- 1 -(4-methy lbenzy l)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexy 1- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 47
[00501] Example 47 is prepared as in Example 45, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 48
Ingredients Amount (mg/unit)
Rimonabant Powder Preparation
10 mg
Rimonabant HCl 2 mg Polyvinylpyrrolidones (PVP) K17 1O mL* Chloroform
Aversive Agent Loading on Sohere
Rimonabant HCl 10 Sugar Spheres (30/35 mesh) 50 Opadry White Y-5-7068 2.5 Ethanol 42.5*
Sphere Overcoating
Opadry White Y-5-7068 3.02 Purified Water 17.1 1 *
Sequestration Overcoating
Eudragit RS30D 12.10 Triethyl Citrate 2.42 Talc 4.84 Purified Water 49.21 *
Final Overcoating
Opadry White Y-5-7068 4.12 Purified Water 23.35*
Total Weight 89.0
* In the final product in negligible quantities only as residual moisture.] Powder Preparation: Dissolve the Rimonabant HCl in chloroform or a suitable alternative organic solvent with mixing. Dissolve PVP in the Rimonabant HCl/chloroform mixture with mixing. Evaporate the chloroform under vacuum, optionally with heating or using a suitable alternative method (e.g., by freeze-drying or by spray-drying) to produce a residual Rimonabant/PVP powder. Micronize the Rimonabant/PVP powder. Aversive Agent Loading on Sphere: Add Rimonabant HC1/PVP in ethanol with stirring, then add Opadry White and continue mixing until a homogenous dispersion is produced. Apply the above dispersion onto the sugar spheres using a fluid bed coater. Sphere Overcoating: Disperse Opadry White in purified water with stirring and apply the dispersion over the sugar spheres loaded with
Rimonabant HCl using a fluid bed coater. Sequestration Overcoating: Mix Eudragit RS30d, triethyl citrate, talc and purified water with stirring and apply the dispersion over the loaded and overcoated sugar spheres using a fluid bed coater (i.e., a fluid bed coating machine). Final Coating: Disperse Opadry White in purified water with stirring and apply the dispersion foregoing (sequestration overcoated product). Cure: Cure the foregoing spheres at 45 ° for approximately 48 to 72 hours.
EXAMPLE 49
[00503] Example 49 is prepared as in Example 48, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxarnide), AM630, SR144528 ([N-[( 1 S)-endo- 1 ,3,3-trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 50
[00504] Example 50 is prepared as in Example 49, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 51
Ingredients Amount (mg/unit)
Aversive Agent Loading on Sphere
Rimonabant HCl 10 Sugar Spheres (30/35 mesh) 50 Opadry White Y-5-7068 2.5 Purified Water 42.5*
Sphere Overcoating
Opadry White Y-5-7068 3.02 Purified Water 17.1 1 *
Sequestration Overcoating
Eudragit RS30D 12.10 Triethyl Citrate 2.42 Talc 4.84 Purified Water 49.21 *
Final Overcoating
Opadry White Y-5-7068 4.12 Purified Water 23.35*
Total Weight 89.0
* In the final product in negligible quantities only as residual moisture.
[00505] Aversive Agent Loading on Sphere: Add Rimonabant HCl in purified water with stirring, then add Opadry White and continue mixing until a homogenous dispersion is produced. Apply the above dispersion onto the sugar spheres using a fluid bed coater. Sphere Overcoating: Disperse Opadry White in purified water with stirring and apply the dispersion over the sugar spheres loaded with rimonabant HCl using a fluid bed coater. Sequestration Overcoating: Mix Eudragit RS30d, triethyl citrate, talc and purified water with stirring and apply the dispersion over the loaded and overcoated sugar spheres using a fluid bed coater (i.e., a fluid bed coating machine). Final Coating: Disperse Opadry White in purified water with stirring and apply the dispersion foregoing (sequestration overcoated product). Cure: Cure the foregoing spheres at 45 ° for approximately 48 to 72 hours.
EXAMPLE 52
[00506] Example 52 is prepared as in Example 51, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5,
0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-l H-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 53
[00507] Example 53 is prepared as in Example 51, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 54

[00508] Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert
installed in fluid-bed equipment. A coating solution is prepared by dissolving the Eudragit RSlOO in ethyl alcohol and dissolving the triethyl citrate in the solution. The Rimonabant HCl cores (700 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed, in a fluid- bed equipped with a Wurster insert.
EXAMPLE 55
[00509] Example 55 is prepared as in Example 54, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl- 1 H-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SRl 41716, SR144528, AM630, HU-308, HU-210, 5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 56
[00510] Example 56 is prepared as in Example 54, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 57 Ingredients Amount (g)
Core Formulation
Rimonabant HCl 70
Methocel E5P 5
Sugar Spheres (16-18 mesh) 700
Ethyl Alcohol (denatured) 200
Purified Water 200
Sequestration Overcoating
Eudragit NE 30D 500
Triethyl Citrate 150
Ethyl Alcohol 1260
[00511] Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Eudragit NE 30D in ethyl alcohol and dissolving the triethyl citrate in the solution. The Rimonabant HCl cores (70 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed in a fluid- bed equipped with a Wurster insert.
EXAMPLE 58
[00512] Example 58 is prepared as in Example 57, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-l H-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichIorophenyl)-3-hexyl-lh-l ,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 59
[00513] Example 59 is prepared as in Example 57, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 60
[00514] Example 60 is prepared as in any of Example 57, Example 58, or
Example 59, except that the Purified water is replaced with 70% ethanol in water.
EXAMPLE 61
Ingredients Amo
Core Formulation
Rimonabant HCl 70
Methocel E5P 5
Sugar Spheres (16-18 mesh) 700
Ethyl Alcohol (denatured) 200
Purified Water 200
Sequestration Overcoating
Ethylcellulose NlO 126
Dibutyl Sebacate 14
Ethyl Alcohol 1260
[00515] Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Ethylcellulose Nl O in ethyl alcohol and dissolving the dibutyl sebacate in the solution. The Rimonabant HCl cores (70 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed in a fluid-bed equipped with a Wurster insert.
EXAMPLE 62
[00516] Example 62 is prepared as in Example 61, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l ,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chlorophenyl)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 63
[00517] Example 63 is prepared as in Example 61, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 64
[00518] Example 64 is prepared as in any of Example 61, Example 62, or
Example 63, except that the Purified water is replaced with 70% ethanol in water.
EXAMPLE 65
Ingredients Amount (g)
Core Formulation
Rimonabant HCl 115
Plasdone K29/32 1 15
Sugar Spheres (18-20 mesh) 1930
Ethyl Alcohol (denatured) 651
Purified Water 651
Sequestration Overcoating
Eudragit RSlOO 189
Dibutyl Sebacate 21
Ethyl Alcohol 2590
[00519] Rimonabant HCl is dissolved in water. An equal volume of denatured alcohol is added. Methocel is added to the above mixture and stirred until it is completely dissolved. The drug layering takes place in a rotor processor insert installed in fluid-bed equipment. A coating solution is prepared by dissolving the Eudragit RSlOO in ethyl alcohol and dissolving the dibutyl sebacate in the solution. The Rimonabant HCl cores (1 15 g) are then coated with the coating solution up to a 20 to 60% weight gain. The coating is performed in a fluid- bed equipped with a Wurster insert.
EXAMPLE 66
[00520] Example 66 is prepared as in Example 65, except that the Rimonabant
HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5-[2,4- dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3- methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes,
polymoφhs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 67
[00521] Example 67 is prepared as in Example 65, except that the Rimonabant
HCI 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 68
[00522] Example 68 is prepared as in any of Example 65, Example 66, or
Example 67, except that the Purified water is replaced with 70% ethanol in water.
EXAMPLE 69
Ingredients Amount (mg)
Rimonabant HCl 5
Sugar Spheres (16-18 mesh) 94.6
Hypromellose 0.4
Ethylcellulose 7.9
Dibutyl Sebacate 1.6
Magnesium Stearate 1.6
[00523] Rimonabant HCl is dispersed in a hydroalcoholic solution of hypromellose with stirring. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of ethylcellulose and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
EXAMPLE 70
Ingredients Amount (nig)
Rimonabant HCl 5
Sugar Spheres (16-18 mesh) 94.6
Hypromellose 0.4
Eudragit RS PO 8.1
Sodium Lauryl Sulfate 0.2
Dibutyl Sebacate 0.8
Magnesium Stearate 2.0
[00524] Rimonabant HCl is dispersed in a hydroalcoholic solution of hypromellose with stirring. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of Eudragit RS, sodium lauryl sulfate and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
EXAMPLE 71
Ingredients Amount (mg)
Rimonabant HCl 5
Sugar Spheres (16-18 mesh) 94.5
Hydroxypropyl cellulose 0.5
Ethylcellulose 9.6
Dibutyl Sebacate 1.5
[00525] Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose using a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of ethylcellulose and dibutyl sebacate in ethanol is prepared, which is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
EXAMPLE 72
Ingredients Amount (mg)
Rimonabant HCl 5 Sugar Spheres (16-18 mesh) 43.8 Hydroxypropyl cellulose 1.3 Ethylcellulose 10 Magnesium Stearate 1
[00526] Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose using a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of ethylcellulose in ethanol is prepared and magnesium stearate is dispersed just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl cores in Glatt GPCG-3 with a 4" Wurster insert.
EXAMPLE 73
Ingredients Amount (mg)
Rimonabant HCl 20
Sugar Spheres (20-25 mesh) 160
Hydroxypropyl cellulose 4
Eudragit RS PO 22
Sodium Lauryl Sulfate 1.2
Dibutyl Sebecate 2.4
Magnesium Stearate 10
[00527] Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose with a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of Eudragit RS, sodium lauryl sulfate and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl in Glatt GPCG-3 with a 4" Wurster insert.
EXAMPLE 74
Ingredients Amount (g)
Rimonabant HCl 5
Sugar Spheres (20-25 mesh) 43.8
Hydroxypropyl cellulose 1.3
Eudragit RS PO 7.4
Sodium Lauryl Sulfate 0.3
Dibutyl Sebecate 0.7
Magnesium Stearate 2.5
[00528] Rimonabant HCl is dispersed in an ethanolic solution of hydroxypropyl cellulose with a mechanical stirrer. The resulting dispersion is layered onto sugar spheres using a rotor granulation process in Glatt GPCG-3 fluid-bed. A polymer solution of Eudragit RS, sodium lauryl sulfate and dibutyl sebacate in ethanol is prepared, and magnesium stearate is dispersed into the polymer solution just prior to spraying. The polymer dispersion is then coated onto Rimonabant HCl in Glatt GPCG-3 with a 4" Wurster insert.
EXAMPLE 75
[00529] Example 75 is prepared as in any of Examples 69, 70, 71, 72, 73 or 74, except that the Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251, AM281 ([N-morpholin-4- yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630, SR144528 ([N-[(l S)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2- yl]5-(4-chloro-3-methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3- carboxamide]), 5-(4-chlorophenyl)-l -(2,4-dichlorophenyl)-3-hexyl- Ih-1, 2,4- triazole, SR141716, SR144528, AM630, HU-308, HU-210, 5-(4- chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 76
[00530] Example 76 is prepared as in any of Examples 69, 70, 71, 72, 73 or 74, except that the Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
[00531] In Example 77 to 99, a substantially non-releasable forms of aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, and alcohol deterrents, or mixtures thereof are prepared by coating aversive agent particles or aversive agent loaded beads with a coating that render it substantially non-releasable.
EXAMPLE 77 Ingredients Amount (mg/unit)
Rimonabant HCl 5
Eudragit RSPO 180
Stearyl alcohol 55
[00532] Stearyl alcohol is passed through an impact mill. The Rimonabant
HCl, Eudragit RSPO and milled stearyl alcohol are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 78
Ingredients Amount (mg/unit)
Rimonabant HCl 20 Eudragit RSPO 180 Stearyl alcohol 30 Stearic acid 30 Butylated hydroxytoluene 2
[00533] Stearyl alcohol is passed through a mill. The Rimonabant HCl,
Eudragit RSPO, milled stearyl alcohol, stearic acid and butylated hydroxytoluene are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 79 Ingredients Amount (mg/unit)
Rimonabant HCl 20
Eudragit RSPO 100
Stearyl alcohol 22
Stearic acid 22
Butylated hydroxytoluene 1.5
[00534] Stearyl alcohol is passed through a mill. The Rimonabant HCl,
Eudragit RSPO, milled stearyl alcohol, stearic acid and butylated hydroxytoluene are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 80
Ingredients Amount (mg/unit)
Rimonabant HCl 5 Eudragit RSPO 90 Stearyl alcohol 15 Stearic acid 15 Butylated hydroxytoluene 1
[00535] Stearyl alcohol is passed through a mill. The Rimonabant HCl,
Eudragit RSPO, milled stearyl alcohol, stearic acid and butylated hydroxytoluene are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 81 Ingredients Amount (mg/unit)
Rimonabant HCl 5
Eudragit RSPO 100
Stearyl alcohol 24
Dibasic calcium phosphate 7
Butylated hydroxytoluene 1.2
[00536] Stearyl alcohol is passed through a mill. The Rimonabant HCl,
Eudragit RSPO, milled stearyl alcohol, dibasic calcium phosphate and butylated hydroxytoluene are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 82
Ingredients Amount (mg/unit)
Rimonabant HCl 3 Eudragit RSPO 90 Stearyl alcohol 22 Dibasic calcium phosphate 6 Butylated hydroxytoluene 1
[00537] Stearyl alcohol is passed through a mill. The Rimonabant HCl,
Eudragit RSPO, milled stearyl alcohol, dibasic calcium phosphate and butylated hydroxytoluene are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 83 Ingredients Amount (mg/unit)
Rimonabant HCl 15
Eudragit RSPO 140
Stearyl alcohol 30
Dibasic calcium phosphate 9
Butylated hydroxytoluene 1.5
[00538] Stearyl alcohol is passed through a mill. The Rimonabant HCl,
Eudragit RSPO, milled stearyl alcohol, dibasic calcium phosphate and butylated hydroxytoluene are mixed in a twin shell blender. The blended material is continuously fed into a twin screw extruder and the resultant strands are collected on a conveyor and allowed to cool. The cooled strands are cut into pellets using a pelletizer. The pellets so formed are screened through a sieve and the desired particle size (sieve portion) is collected.
EXAMPLE 84 Step Ingredients Amount (mg/unit)
Rimonabant HCl 10
Non-pareil beads (30-35 mesh) 200
Opadry Clear (HPMC) 2
Sodium ascorbate 0.13
Ascorbic Acid 0.25
2 Eudragit L30D 1 1 Triethyl citrate 2.1 Cabosil 0.55
3 Eudragit RS30D 88 Triethyl citrate 17.5 Cabosil 4.4
4 Opadry Clear (HPMC) 9.5 Cabosil 1.4
[00539] (1) The Rimonabant HCl, ascorbic acid, sodium ascorbate and Opadry
Clear is dissolved in water. The resultant solution is sprayed on non-pareil beads in a fluid bed coater with Wurster insert. (2) The Eudragit L30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the drug-loaded beads in a fluid bed coater. (3) The Eudragit RS30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the beads in a fluid bed coater (controlled release coat). (4) The Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours.
EXAMPLE 85 Step Ingredients Amount (mg/unit)
Rimonabant HCl 10 Non-pareil beads (30-35 mesh) 100 Opadry Clear (HPMC) 1 Sodium ascorbate 0.6 Ascorbic Acid 0.1
Eudragit L30D 5 Triethyl citrate 1 Cabosil 0.25
Eudragit RS30D 40 Triethyl citrate 8 Cabosil 2
Opadry Clear (HPMC) 4 Cabosil 0.7
[00540] (1) The Rimonabant HCl, ascorbic acid, sodium ascorbate and Opadry
Clear is dissolved in water. The resultant solution is sprayed on non-pareil beads in a fluid bed coater with Wurster insert. (2) The Eudragit L30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion
is sprayed onto the drug-loaded beads in a fluid bed coater. (3) The Eudragit RS30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the beads in a fluid bed coater (controlled release coat). (4) The Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours.
EXAMPLE 86
Step Ingredients Amount (mg/unit)
1 Rimonabant HCl 3 Non-pareil beads (30-35 mesh) 40 Opadry Clear (HPMC) 0.4 Sodium ascorbate 0.03 Ascorbic Acid 0.05
2 Eudragit L30D 2.2 Triethyl citrate 0.5 Cabosil 0.12
3 Eudragit RS30D 19 Triethyl citrate 3.5 Cabosil 0.9
4 Opadry Clear (HPMC) 1.9 Cabosil 0.28 ] (1) The Rimonabant HCl, ascorbic acid, sodium ascorbate and Opadry
Clear is dissolved in water. The resultant solution is sprayed on non-pareil beads in a fluid bed coater with Wurster insert. (2) The Eudragit L30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the drug-loaded beads in a fluid bed coater. (3) The Eudragit RS30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the beads in a fluid bed coater (controlled release coat). (4) The Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours.
EXAMPLE 87 Step Ingredients Amount (mg/unit)
1 Rimonabant HCl 1
Non-pareil beads (30-35 mesh) 68
Opadry Clear (HPMC) 0.55
2 Eudragit L 2.6 Triethyl citrate 0.65 Glyceryl monostearate 0.24
3 Eudragit RS30D 44 Triethyl citrate 8.8 Cabosil 2.2
4 Opadry Clear (HPMC) 2.1 Cabosil 1.4
(1) The Rimonabant HCl, ascorbic acid and Opadry Clear is dissolved in water. The resultant solution is sprayed on non-pareil beads in a fluid bed coater with Wurster insert. (2) The Eudragit L30D, triethyl citrate and glyceryl monostearate are dispersed in water and the resulting dispersion is sprayed onto the drug-loaded beads in a fluid bed coater. (3) The Eudragit RS30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the beads in a fluid bed coater (controlled release coat). (4) The Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours. The resulting bead formulation can be incorporated into a controlled release Opioid Agonist granulation and the mixture compressed into tablets.
EXAMPLE 88
Step Ingredients Amount (mg/unit)
1 Rimonabant HCl 5
Non-pareil beads (30-35 mesh) 120 Opadry Clear (HPMC) 1
2 Eudragit L 5 Triethyl citrate 1.2 Glyceryl monostearate 0.5
3 Eudragit RS30D 80 Triethyl citrate 15 Cabosil 5
4 Opadry Clear (HPMC) 4 Cabosil 2
[00543] (1) The Rimonabant HCl, ascorbic acid and Opadry Clear is dissolved in water. The resultant solution is sprayed on non-pareil beads in a fluid bed coater with Wurster insert. (2) The Eudragit L30D, triethyl citrate and glyceryl monostearate are dispersed in water and the resulting dispersion is sprayed onto the drug-loaded beads in a fluid bed coater. (3) The Eudragit RS30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the beads in a fluid bed coater (controlled release coat). (4) The Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours. The resulting bead formulation can be incorporated into a controlled release Any of example 1 to 99 granulation and the mixture compressed into tablets.
EXAMPLE 89
Step Ingredients Amount (mg/unit)
1 Rimonabant HCl 10 Non-pareil beads (30-35 mesh) 120 Opadry Clear (HPMC) 4
2 Eudragit L 10 Triethyl citrate 2.5 Glyceryl monostearate 1
3 Eudragit RS30D 120 Triethyl citrate 25 Cabosil 7
4 Opadry Clear (HPMC) 8 Cabosil 4.5
[00544] (1) The Rimonabant HCl, ascorbic acid and Opadry Clear is dissolved in water. The resultant solution is sprayed on non-pareil beads in a fluid bed coater with Wurster insert. (2) The Eudragit L30D, triethyl citrate and
glyceryl monostearate are dispersed in water and the resulting dispersion is sprayed onto the drug-loaded beads in a fluid bed coater. (3) The Eudragit RS30D, triethyl citrate and Cabosil are dispersed in water and the resulting dispersion is sprayed onto the beads in a fluid bed coater (controlled release coat). (4) The Opadry Clear is dissolved in water and the resulting solution sprayed onto beads in a fluid bed coater. The coated beads are then cured at 60° C for 24 hours. The resulting bead formulation can be incorporated into a controlled release Any of example 1 to 99 granulation and the mixture compressed into tablets.
EXAMPLE 90 Ingredients Amount (mg/unit)
Rimonabant HCl 2
Eudragit RSPO 88
Stearyl alcohol 15
Stearic acid 15
Butylated hydroxytoluene 1
[00545] The stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender. The mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer. The Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar, the Conveyor is set to provide extrudate diameter of 1 mm and the Pelletizer is set to provide pellets of approximately 1 mm length. Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
EXAMPLE 91 Ingredients Amount (mg/unit)
Rimonabant HCl 5
Eudragit RSPO 88
Stearyl alcohol 15
Stearic acid 15
Butylated hydroxytoluene 1
[00546] The stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender. The
mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer. The Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar, the Conveyor is set to provide extrudate diameter of 1 mm and the Pelletizer is set to provide pellets of approximately 1 mm length. Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
EXAMPLE 92
Ingredients Amount (mg/unit)
Rimonabant HCl 10
Eudragit RSPO 150
Stearyl alcohol 20
Stearic acid 20
Butylated hydroxytoluene 1.5
[00547] The stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender. The mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer. The Powder Feeder rate is set at approximately 4.2 kg/hr and a vacuum of 980 mBar, the Conveyor is set to provide extrudate diameter of 1 mm and the Pelletizer is set to provide pellets of approximately 1 mm length. Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
EXAMPLE 93
Ingredients Amount (mg/unit)
Rimonabant HCl 10
Eudragit RSPO 400
Stearyl alcohol 25
Stearic acid 25
Butylated hydroxytoluene 2.5
[00548] The stearic acid, stearyl alcohol, Rimonabant HCl, Butylated hydroxytoluene and Eudragit RSPO are blended using a V-blender. The mixture is extruded using a Powder Feeder Melt extruder (equipped with the 6x1 die head), Conveyor, Lasermike and Pelletizer. The Powder Feeder rate is
set at approximately 4.2 kg/hr and a vacuum of 980 mBar, the Conveyor is set to provide extrudate diameter of 1 mm and the Pelletizer is set to provide pellets of approximately 1 mm length. Pellets are screened using #16 mesh and #20 mesh screens and material retained between #16 and #20 mesh screen is retained.
EXAMPLE 94
[00549] Examples 94 is prepared in accordance to any of Examples 77 to 93, except that the Rimonabant is first prepared as a powder using the method of Example 48.
EXAMPLE 95
[00550] Examples 95 is prepared in accordance to any of Examples 77 to 89, except that in the first step, the dissolution is in a hydroalcoholic solution.
EXAMPLE 96
[00551] Examples 96 is prepared in accordance to any of Examples 77 to 89, except that in the first step, the dissolution is in denatured ethyl alcohol.
EXAMPLE 97
[00552] Examples 97 is prepared in accordance to any of Examples 77 to 89, except that in the first step, the dissolution is in a suitable organic solvent.
EXAMPLE 98
[00553] Example 98 is prepared as in any of Examples 77 to 83, except that the
Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 0.25, 0.5, 0.75, 1.0, 1.5 or 2.0 times the quantity of Rimonabant HCl, selected from the group comprising AM251 , AM281 ([N-morpholin-4-yl]-5-[2,4-yl]-5- [2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3-carboxamide), AM630,
SR144528 ([N-[(lS)-endo-l,3,3-trimethylbicyclo(2.2.1)heptan-2-yl]5-(4- chloro-3-methyl-phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichloropheny l)-3-hexyl- 1 h- 1 ,2,4-triazole, SR 141716, SR144528, AM630, HU-308, HU-210, 5-(4-chlorophenyl)-l -(2,4- dichlorophenyl)-3-hexyl-lh-l,2,4-triazole, and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
EXAMPLE 99
[00554] Example 99 is prepared as in any of Examples 77 to 83, except that the
Rimonabant HCl 10 mg is replaced with an amount of aversive agent equal to 1.0, 1.5, 2.0, 3.0, 4.0, or 6.0 times the quantity of Rimonabant HCl, selected from the group comprising Disulfiram, Calcium Carbimide and or their pharmaceutically acceptable salts, prodrugs, esters, analogs, derivatives, solvates, complexes, polymorphs, hydrates and metabolites, as racemates or an individual diastereoisomers or enantiomeric isomers thereof or mixtures thereof.
[00555] In Example 100 to 1 16, tablet formulation comprising an immediate release opioid agonist with a suitable amount of substantially non-releasable aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents and mixtures thereof is prepared. The preparation of substantially non-releasable aversive agent forms has been described in the forgoing examples. The amount of substantially non- releasable aversive agent added to the dosage forms of Example 100 to 1 16 will vary based on a variety of factors previously described.
EXAMPLE 100
Ingredients Amount/Unit
1 2 mg
Hydromorphone
2 7.5 mg
Polyvinylpyrrolidine
3 Lactose 30 mg
4 Alcohol SD3A-2 proof 3 Ml
5 Stearic acid 5 mg
6 Talc 7.5 mg
7 Cornstarch 20 mg
8 Any of example 1 to 99 Suitable Amount
[00556] Blend 1, 2 and 3 together; pass through a 40-mesh screen. Add 4 slowly and knead well. Screen wet mass through a 4-mesh screen. Dry the granulation at 50 0C overnight. Screen the dried granulation through a 20-
mesh screen. Bolt 5, 6 and 7 through a 60-mesh screen prior to mixing by tumbling with granulation. Tumble and mix contents with 8 and compress using a tablet punch. Film coat as desired.
[00557] In some embodiments, the compressed tablets can be formulated using conventional dry granulation procedures and equipment. In other embodiments, the uncompressed granulate can be filled in a hard shell capsule.
EXAMPLE 101
[00558] See procedure outlined in Example 100.
Ingredients Amount/Unit
1 Levorphanol base 2.5 mg
2 Polyvinylpyrrolidine 7.5 mg
3 30 mg
Lactose
4 Alcohol SD3A-2 proof 3 Ml
5 Stearic acid 5 mg
6 7.5 mg
Talc
7 Cornstarch 20 mg
8 Any of example 1 to 99 Suitable Amount
EXAMPLE 821
[00559] See procedure outlined in Example 100.
Ingredients Amount/Unit
1 Morphine base 25 mg
2 7.5 mg
Polyvinylpyrrolidine
3 Lactose 30 mg
4 Alcohol SD3A-2 proof 3 Ml
5 Stearic acid 5 mg
6 Talc 7.5 mg
7 Cornstarch 20 mg
8 Any of example 1 to 99 Suitable Amount
EXAMPLE 102
[00560] See procedure outlined in Example 100.
Ingredients Amount/Unit
1 Meperidine HCl 100 mg
2 7.5 mg
Polyvinylpyrrolidine
3 30 mg
Lactose
4 Alcohol SD3A-2 proof 3 Ml
5 5 mg
Stearic acid
6 7.5 mg
Talc
7 Cornstarch 20 mg
8 Any of example 1 to 99 Suitable Amount
EXAMPLE 103
[00561] See procedure outlined in Example 100.
Ingredients Amount/Unit
1 Propiram HCl 25 mg
2 7.5 mg
Polyvinylpyrrolidine
3 Lactose 30 mg
4 Alcohol SD3A-2 proof 3 Ml
5 Stearic acid 5 mg
6 7.5 mg
Talc
7 Cornstarch 20 mg
8 Any of example 1 to 99 Suitable Amount
EXAMPLE 104
[00562] See procedure outlined in Example 100.
Ingredients Amount/Unit
1 Codeine HCl 100 mg
2 7.5 mg
Polyvinylpyrrolidine
3 30 mg
Lactose
4 Alcohol SD3A-2 proof 3 Ml
5 5 mg
Stearic acid
6 7.5 mg
Talc
7 20 mg
Cornstarch
8 Any of example 1 to 99 Suitable Amount
EXAMPLE 105
Ingredients Amount/Unit
1 2 mg
Levorphanol tartrate
2 Lactose (granular, 12 mesh) 25 mg
3 20 mg
Starch
4 20 mg
Talc
5 0.3 mg
Magnesium Stearate
6 Any of example 1 to 99 Suitable Amount
[00563] Mix ingredients 1 to 5 thoroughly. Compress into slugs. Grind and screen to 14- to 16-mesh granules. Mix thoroughly with 6. Compress into tablets using a punch. Film coat as desired. In some embodiments, the uncompressed granulate can be filled in a hard shell capsule.
EXAMPLE 106
Ingredients Amount/Unit
1 Oxycodone HCl 2.5 mg
2 25 mg
Lactose (granular, 12 mesh)
3 Starch 20 mg
4 Talc 20 mg
5 Magnesium Stearate 0.3 mg
6 Any of example 1 to 99 Suitable Amount
[00564] See procedure outlined in Example 105.
EXAMPLE 107
Ingredients Amount/Unit
1 Moφhine sulfate 25 mg
2 Lactose (granular, 12 mesh) 50 mg
3 Starch 30 mg
4 Talc 25 mg
5 Magnesium Stearate 0.5 mg
6 Any of example 1 to 99 Suitable Amount
[00565] See procedure outlined in Example 105.
EXAMPLE 108
Ingredients Amount/Unit
1 Prop i ram HCl 100 mg
2 100 mg
Lactose (granular, 12 mesh)
3 Starch 40 mg
4 Talc 35 mg
5 Magnesium Stearate 0.8 mg
6 Any of example 1 to 99 Suitable Amount
[00566] See procedure outlined in Example 105.
EXAMPLE 109
Ingredients Amount/Unit
1 Morphine 25 mg
2 50 mg
Lactose (granular, 12 mesh)
3 30 mg
Starch
4 Talc 25 mg
5 Magnesium Stearate 0.5 mg
6 Any of example 1 to 99 Suitable Amount
[00567] See procedure outlined in Example 105.
EXAMPLE 1 10
Ingredients Amount/Unit
1 Hydrocodone 10 mg
2 100 mg
Lactose (granular, 12 mesh)
3 40 mg
Starch
4 35 mg
Talc
5 0.8 mg
Magnesium Stearate
6 Any of example 1 to 99 Suitable Amount
[00568] See procedure outlined in Example 105.
EXAMPLE 1 1 1
Ingredients Amount/Unit
1 Oxycodone 5 mg
2 HPMC 2208, USP 100 mg
3 Carnauba wax
15 mg
4 HPMC 2910, USP 12 mg
5 Magnesium Stearate
2 mg
6 Stearic acid 6 mg
7 Talc
2 mg
8 Any of example 1 to 99 Suitable Amount
Place the ingredients 1, 2 and 3 in the granulator and mix for 15 minutes. Dissolve ingredient 4 in water (mix in hot water, then cool down) and spay into the fluidized mixture. Dry to approximately 5% moisture. Sequentially add ingredient 5, 6 and 7, with mixing steps between each addition. Thoroughly mix contents with 8. Compress into tablets using a punch. Film coat as desired. In some embodiments, the uncompressed granulate can be filled in a hard shell capsule.
EXAMPLE 1 12
Ingredients Amount/Unit
1 Hydromoφhone base
4 mg
2 HPMC 2208, USP
150 mg
3 Carnauba wax
30 mg
4 HPMC 2910, USP
15 mg
5 Magnesium Stearate
2 mg
6 Stearic acid
8 mg
7 Talc
3 mg
8 Any of example 1 to 99 Suitable Amount
[00570] See procedure outlined in Example 1 1 1.
EXAMPLE 1 13
Ingredients Amount/Unit
1 Oxymorphone HCl
30 mg
2 HPMC 2208, USP
150 mg
3 Carnauba wax
30 mg
4 HPMC 2910, USP
15 mg
5 Magnesium Stearate
2 mg
6 Stearic acid
8 mg
7 Talc
3 mg
8 Any of example 1 to 99 Suitable Amount
[00571] See procedure outlined in Example 1 11.
EXAMPLE 1 14
Ingredients Amount/Unit
1 Codeine Sulfate 100 mg
2 HPMC 2208, USP
150 mg
3 Carnauba wax
30 mg
4 HPMC 2910, USP
15 mg
5 Magnesium Stearate
2 mg
6 Stearic acid
8 mg
7 Talc
3 mg
8 Any of example 1 to 99 Suitable Amount
[00572] See procedure outlined in Example 1 11.
EXAMPLE 1 15
Ingredients Amount/Unit
1 Oxycodone HCl 10 mg
2 HPMC 2208, USP 100 mg
3 Carnauba wax 15 mg
4 HPMC 2910, USP 10 mg
5 Magnesium Stearate 1.25 mg
6 Stearic acid 5 mg
7 Talc 1.5 mg
8 Any of example 1 to 99 Suitable Amount
[00573] See procedure outlined in Example 1 1 1.
EXAMPLE 116
Ingredients Amount/Unit
1 Hydromorphone HCl
8 mg
2 HPMC 2208, USP
75 mg
3 Carnauba wax 15 mg
4 HPMC 2910, USP
7.5 mg
5 Magnesium Stearate
1 mg
6 Stearic acid
4 mg
7 Talc
1.5 mg
8 Any of example 1 to 99 Suitable Amount [00574] See procedure outlined in Example 1 1 1.
[00575] A wide variety of methods are described in the art and her preparation of controlled release dosage forms (e.g., tablets and capsule forms) of pharmaceuticals.
[00576] The releasable or substantially releasable Any of example 1 to 99 in controlled release form is generally prepared separately from the substantially non-releasable aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents, and mixtures thereof.
The final dosage form containing the releasable or substantially releasable Any of example 1 to 99 form and the substantially non-releasable aversive agent form is generally prepared by mixing the two forms and compressing it into a tablet and film coating as desired or filling into a capsule. [00577] Examples 1 17 to 1 19 are controlled release formulations of releasable or substantially releasable opioid agonists.
EXAMPLE 1 17
Ingredients Amt/Unit (mg)
Oxycodone HCl 30
Spray Dried Lactose 60
Povidone 5
Eudragit RS30D (solids) 10
Triacetin 2
Stearyl Alcohol 25
Talc 2.5
Magnesium Stearate 1.25
Opadry Pink Y-S-14518A 4.0
[00578] 1. Granulation: Spray the Eudragit/Triacetin dispersion onto the
Oxycodone, Spray Dried Lactose and Povidone using a fluid bed granulator. 2. Milling: Discharge the granulation and pass through a mill. 3. Waxing: Melt the stearyl alcohol and add to the milled granulation using a mixer. Allow to cool. 4. Milling: Pass the cooled granulation through a mill. 5. Lubrication: Lubricate the granulation with talc and magnesium stearate using a mixer. 6. Compression: Compress the granulation into tablets using a tablet press. 7. Film coating: Apply an aqueous film coat to the tablets.
EXAMPLE 1 18 Ingredients Amt/Unit (mg)
Moφhine sulfate 30
Eudragit RSPO 76
Eudragit RLPO 4
Stearyl Alcohol 25
[00579] 1. Blend milled Stearyl Alcohol, Eudragit RLPO, Morphine sulfate, and Eudragit RSPO using a Hobart Mixer. 2. Extrude the granulation using a
Powder Feeder, Melt Extruder (equipped with the ό.times.l mm die head), Conveyor, Lasermike, and Pelletizer. Powder feed rate-40 g/min; vacuum- .about 980 mBar; Conveyor, such that diameter of extrudate is 1 mm , Pelletizer, such that pellets are cut to 1 mm in length. Screen pellets using #16 mesh and #20 mesh screens. Collect material that passes through the #16 mesh screen and is retained on the #20 mesh screen. 4. Fill capsules with the pellets.
EXAMPLE 1 18b Ingredients Amt/Unit (mg)
Hydromorphone HCl 12
Eudragit RSPO 77
Ethocel 4.5
Stearic acid 27
[00580] 1. Blend milled Stearic acid, ethocel, Hydromorphone HCl, and
Eudragit RSPO using a V-blender. 2. Extrude the mixture using a Powder Feeder, Melt Extruder'(equipped with the ό.times.l mm die head), Conveyor, Lasermike, and Pelletizer. Powder feed rate, 1.2 kg/hr; vacuum, about.980 mBar; Conveyor, such that diameter of extrudate is 1 mm; Pelletizer, such that pellets are cut to 1 mm in length. 3. Screen pellets using #16 mesh and #20 mesh screens. Collect material that passes through the #16 mesh screen and is retained on the #20 mesh screen. Fill pellets in capsules.
EXAMPLE 1 19 Steps Ingredients Amt/unit (mg)
Levoφhanol tartrate 12
Non-pareil beads (30/35 mesh) 45 Opadry Clear 2.5
2 Eudragit RS3-D (dry) 7.2 Eudragit RL30D (dry) Q 4 Triethyl citrate Cabosil
3 Opadry Clear (HPMC) 1.9 Cabosil 0.28
[00581] 1. Dissolve Levoφhanol tartrate and Opadry (HPMC) in water. Spray the drug solution onto nonpareil beads in a fluid bed coater with Wurster insert. 2. Disperse Eudragit RS, Eudragit RL, triethyl citrate, and Cabosil in water. Spray the dispersion onto the beads in the fluid bed coater. 3. Dissolve
Opadry in water. Spray the solution onto the beads in the fluid bed coater. 4. Cure the beads at 6O.degree. C. for 24 hours.
EXAMPLE 120
[00582] The substantially non-releasable aversive agent forms described in any of Examples 1 to 99 can be combined with the releasable or substantially releasable opioid agonists in controlled release form of any of Examples 1 17 to 1 1 1 to provide a releasable or substantially releasable opioid agonist controlled release dosage form with an a substantially non-releasable aversive agent (i.e., sequestered agent). The dosage form may be compressed into a tablet or filled into a capsule.
[00583] Examples 121 to 134 are controlled release formulations of releasable or substantially releasable opioid agonists with substantially non-releasable aversive agent (i.e., sequestered agent) selected from the group comprising cannabinoid antagonists, alcohol deterrents and mixtures therof. The opioid agonist in controlled release form is generally prepared separately from the substantially non-releasable aversive agent. The final dosage form is generally prepared by mixing the two forms and compressing it into a tablet and film coating as desired or filling into a capsule.
EXAMPLE 121

♦Remains in product as residual moisture only.
[00584] Examples 121 to 125: 1. Pass the Stearyl Alcohol flakes through an oscillating mill. 2. Mix the specified opioid agonist, milled Stearyl Alcohol, Anhydrous Dicalcium Phosphate, Microcrystalline Cellulose, and Glyceryl
Behenate in a twin shell blender. 3. Continuously feed the blended material into a twin screw extruder and collect the resultant heated material on a conveyor. 4. Allow the extrudate to cool on the conveyor. 5. Mill the cooled extrudate using an oscillating mill. 6. Blend the milled extrudate, substantially non-releasable aversive agent beads (selected from Example 1 to 99), and Magnesium Stearate. 7. Compress the resultant granulation using a tablet press, preferably into a caplet. 8. Prepare a film coating solution by dispersing the Opadry in Purified Water and applying it to the tablet.

*Remains in product as residual moisture only.

* Remains in product as residual moisture only.
EXAMPLE 124

*Remains in product as residual moisture only.

*Remains in product as residual moisture only. ] Examples 126 to 128: Plasticize the Eudragit with Triacetin by mixing. 2. Place the specified Opioid Agonist, Spray Dried Lactose, and Povidone into a fluid bed granulator and apply the above solution. 3. Pass the granulation through a rotating impeller mill. 4. Dry granulation if moisture content is too high. 5. Melt Stearyl Alcohol and wax the above granulation by adding melted Stearyl Alcohol onto granulation while mixing. 6. Cool the waxed granulation in a fluid bed dryer. 7. Pass the cooled waxed granulation through a rotating impeller mill. 8. Blend the milled waxed granulation, Talc, Magnesium Stearate, and substantially non-releasable aversive agent beads (selected from Example 1 to 99). 9. Compress the resultant granulation using a
tablet press. 10. Prepare a film coating solution by dispersing the Opadry in Purified Water and applying it to the tablet.

* Remains in product as residual moisture only.

*Remains in product as residual moisture only.
EXAMPLE 128


♦Remains in product as residual moisture only.
EXAMPLE 129
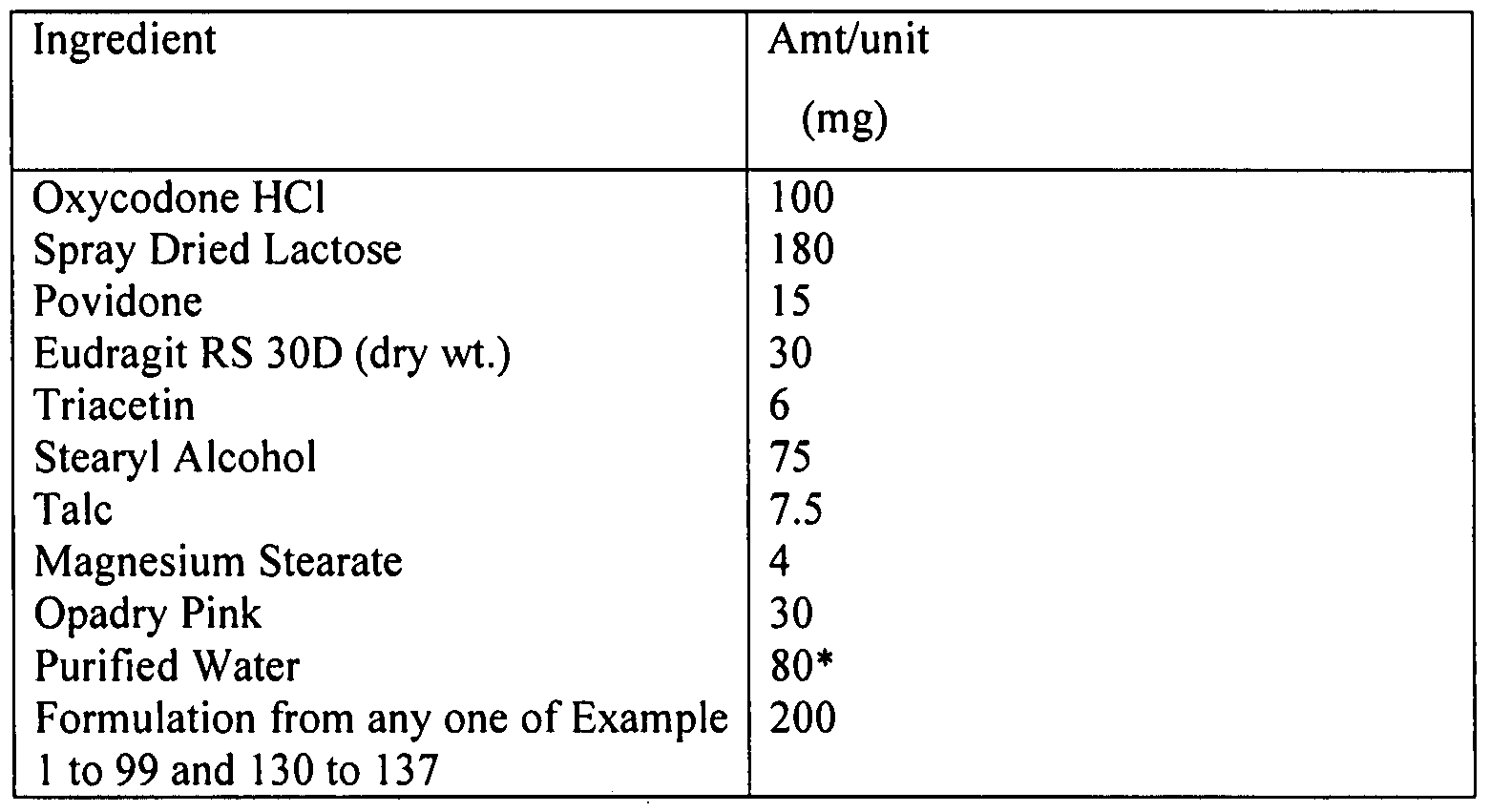
♦Remains in product as residual moisture only. ] Examples! 30 to 134: 1. Pass Stearyl Alcohol flakes through an impact mill. 2. Mix specified Opioid Agonist, Eudragit, Ethylcellulose and milled Stearyl Alcohol in a twin shell blender. 3. Continuously feed the blended material into a twin screw extruder and collect the resultant strands on a conveyor. 4. Allow the strands to cool on the conveyor. 5. Cut the cooled strands into pellets using a Pelletizer. 6. Screen the pellets and collect desired sieve portion. 7. Fill the extruded Opioid Agonist pellets and the substantially non-releasable aversive agent beads (selected from any of Examples 1 to 99) into capsules.
EXAMPLE 130
Ingredient Amt/unit (mg)
Hydromoφhone base 15
Eudragit RSPO 76.5
Ethylcellulose 4.5
Stearyl Alcohol 27
Formulation from any one of Example 240
1 to 99 and 130 to 137
EXAMPLE 131

EXAMPLE 132
Ingredient Amt/unit (mg)
Morphine sulfate 50
Eudragit RSPO 300
Ethylcellulose 18
Stearyl Alcohol 108
Formulation from any one of Example 200
I to 99 and l30 to l37
EXAMPLE 133
Ingredient Amt/unit (mg)
Tramadol HCl 100
Eudragit RSPO 300
Ethylcellulose 18
Stearyl Alcohol 108
Formulation from any one of Example 200
1 to 99 and 130 to 137
EXAMPLE 134
Ingredient Amt/unit (mg)
Propiram HCl 100
Eudragit RSPO 200
Ethylcellulose 12
Stearyl Alcohol 65
Formulation from any one of Example 200
1 to 99 and 130 to 137
EXAMPLE 135
[00587] The cannabinoid antagonists of any of Examples 1 to 99 is first converted to a solid form and micronized using methods know in the art and then incorporated into the method and process of Examples 1 to 99.
EXAMPLE 136
[00588] The cannabinoid antagonists selected from the group consisting of SR
141716A [Rimonabant or N-piperidino-5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-4-methyl-3-pyrazole-carboxamide]), AM251, AM 281 ([N- moφholin-4-yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl-l H-pyrazole-3- carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l,3,3- trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3-methyl-phenyl)-l-(4- methylbenzyl)pyrazole-3-carboxamide]), 5-(4-chloropheny I)-I -(2,4- dichloropheny l)-3-hexy 1- 1 h- 1 ,2,4-triazole, 8-chloro- 1 -(2',4'-dichlorophenyl)- N-piperidin-l-yl-l^Sjό-tetrahydrobenzotόJJcycloheptaP^-ctøyrazole-S- carboxamide 4a, pyrazole class cannabinoid antagonists (e.g., SR141716 and SR144528), aminoalkylindole class cannabinoid antagonists (e.g., AM630), imidazolinedione class cannabinoid antagonists and triazole class cannabinoid antagonists, pyridone derivative class cannabinoid antagonists, quinolone derivative class cannabinoid antagonists, tricyclic derivatives of 1- benzylpyrazole-3-carboxylic acid, HU-308, HU-210, cannabidiol, tricyclic pyrazoles, analogs of 8-chloro-l-(2',4'-dichlorophenyl)-N-piperidin-l-yl- l ,4,5,6-tetrahydrobenzo[6,7]cyclohepta[l,2-c]pyrazole-3-carboxamide, and 5- (4-chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-lh-l,2,4-triazole is prepared according to methods know in the art (e.g., In: Water Insoluble Drug Formulations, Rong Liu (Ed.), Taylor & Francis, CRC Press, 2000, Boca Raton, FL) and compressed into a tablet or filled uncompressed into a capsule.
EXAMPLE 137
[00589] The cannabinoid antagonists selected from the group consisting of SR
141716A [Rimonabant or N-piperidino-5-(4-chloropheny I)-I -(2,4- dichlorophenyl)-4-methyl-3-pyrazole-carboxamide]), AM251, AM 281 ([N- moφholin-4-yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl-lH-pyrazole-3-
carboxamide), AM630, SR 144528 ([N-[(l S)-endo-l,3,3- trimethylbicyclo(2.2.1)heptan-2-yl]5-(4-chloro-3-methyl-phenyl)-l-(4- methylbenzyl)pyrazole-3-carboxamide]), 5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-3-hexyl-lh-l ,2,4-triazole, 8-chloro-l-(2',4'-dichlorophenyl)- N-piperidin-l-yl-l ^jSjό-tetrahydrobenzofόJlcycloheptatl^-clpyrazole-S- carboxamide 4a, pyrazole class cannabinoid antagonists (e.g., SR141716 and SRl 44528), aminoalkylindole class cannabinoid antagonists (e.g., AM630), imidazolinedione class cannabinoid antagonists and triazole class cannabinoid antagonists, pyridone derivative class cannabinoid antagonists, quinolone derivative class cannabinoid antagonists, tricyclic derivatives of 1- benzylpyrazole-3-carboxylic acid, HU-308, HU-210, cannabidiol, tricyclic pyrazoles, analogs of 8-chloro-l-(2',4'-dichlorophenyl)-N-piperidin-l-yl- l,4,5,6-tetrahydrobenzo[6,7]cyclohepta[l,2-c]pyrazole-3-carboxamide, and 5- (4-chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-l h-l ,2,4-triazole is prepared according to methods known in the art (see U.S. patent No. 7,229,641, 7,223,770, 7, 193,084, 7,186,754, 7,138, 1 13, 7, 125,568, 7, 1 12,340, 7, 105,685, 7,060,263, 7,037,528, 7,026,290, 6,991,809, 6,977,085, 6,951,656, 6,942,856, 6,923,988, 6,890,549, 6,884,436, 6,869,617, 6,867,024, 6,864,231, 6,835,396, 6,825,179, 6,806,069, 6,790,460, 6,730,330, 6,683,194, 6,683,102, 6,630,161 , 6,623,734, 6,589,955, 6,589,562, 6,589,556, 6,569,463, 6,565,873, 6,544,646, 6,541 ,030, 6,461,634, 6,407,128, 6,406,717, 6,403, 1 16, 6,375,982, 6,348,506, 6,248,363, 6, 191 , 172, 6,045,826, 5,989,583, 5,891 ,469.
[00590] All of patents and patent applications cited herein are incorporated by reference in their entirety.
[00591] All of publications and references cited herein are incorporated by reference in their entirety.
[00592] Various immediate release and controlled release dosage forms comprising releasable or substantially releasable cannabinoid agonists and non-releasable and substantially non-releasable aversive agents selected from the group comprising cannabinoid antagonists, alcohol deterrents and mixtures therof may be prepared using the pharmaceutical compositions and methods of the invention.
Having now fully described the invention, those of ordinary skill in the art can understand that the same can be performed within a wide and equivalent range of conditions, formulations, and other parameters without affecting the scope of the invention or any embodiment thereof.
Claims
1. An oral dosage form having a reduced potential for abuse, misuse, tampering, and toxicity upon tampering, comprising: (a) a releasable opioid agonist; (b) an aversive agent which is sequestered in the intact dosage form but being releasable upon tampering of said dosage form, the aversive agent when released upon tampering of said dosage form at least partially blocking the effect of the opioid agonist and/or at least partially blocking the effect of another abusable drug not included in the dosage form.
2. An oral dosage form comprising: (a) an opioid agonist; (b) an aversive agent which is sequestered in the intact dosage form but being releasable upon tampering of said dosage form, such that the intact dosage form releases 49% or less, preferably 42% or less, and more preferably 36% or less, 24.6% or less, 10% or less, or 6.2% or less of the aversive agent after 36 hours based on the in-vitro dissolution of the dosage form in 900 ml of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm and 370C with a switch to Simulated Intestinal Fluid at 1 hour, the aversive agent when released upon tampering of said dosage form at least partially blocking the effect of the opioid agonist and/or at least partially blocking the effect of another abusable drug not included in the dosage form.
3. The oral dosage form of claim 2, wherein the intact dosage form releases preferably 42% or less, and more preferably 36% or less, 24.6% or less, 10% or less, or 6.2% or less of the aversive agent after 36 hours based on the in-vitro dissolution of the dosage form in 900 ml of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm and 37°C with a switch to Simulated Intestinal Fluid at 1 hour.
4. An oral dosage form, comprising (a) an opioid agonist in releasable form; (b) an aversive agent in substantially non-releasable form from the intact dosage form but being releasable upon tampering
of said dosage form, such that the ratio of the mean Cmax of the aversive agent after single dose oral administration of the dosage form after tampering to the mean Cmax of aversive agent after single dose oral administration of an intact dosage form is at least 1.5:1.
5. The oral dosage form of claim 4 wherein said ratio is at least 3: 1 ; or at least 6: 1 ; or at least 10: 1 ; or at least 20: 1 ; or at least 30: 1 ; or at least 40: 1 ; or at least 50:1 ; or at least 70: 1 ; or at least 100: 1 ; or at least 500: 1.
6. The oral dosage form of claim 2, wherein the dissolution rate is measured using hen measured by a USP Type II (paddle) apparatus at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C.
7. The oral dosage form of any of claims 1-5, wherein the ratio of the amount of aversive agent released from the dosage form after tampering to the amount of the aversive agent released from the intact dosage form is about 4: 1 or greater, based on the in-vitro dissolution at 1 hour of the dosage form in 900 mL of Simulated Gastric Fluid using a USP Type II (paddle) apparatus at 75 rpm at 37 degrees C.
8. The oral dosage form of claim 7, wherein the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 2 hour is about 4: 1 or greater.
9. The oral dosage form of claim 7, wherein the ratio of the amount of aversive agent contained in the intact dosage form to the amount of the aversive agent released from the intact dosage form after 3, 4, 6, 8, 12 or 24 hours is about 4:1 or greater.
10. The oral dosage form of any of claims 1-7, wherein the opioid agonist is in sustained release form.
1 1. The oral dosage form of claim 10, which is suitable for twice-a- day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by
the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 47.5% at 1 hour, from about 10% to about 65% at 2 hours, from about 15% to about 70% at 4 hours, from about 25% to about 77.5% at 6 hours, from about 35% to about 87.5% at 9 hours, and greater than about 65% at 12 hours.
12. The oral dosage form of claim 1 1, providing an in-vitro release rate by weight of opioid agonist between 0% and about 60% at 1 hour, between about 0% and about 80% at 2 hours, between about 2% and about 95% at 4 hours and between about 10% and about 100% at 8 hours and greater than 60% at 12 hours.
13. The oral dosage form of claim 10, which is suitable for once-a- day administration to a human patient; said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of from 0% to about 30% at 1 hour, from about 10% to about 65% at 4 hours, from about 20% to about 70% at 8 hours, from about 25% to about 80% at 12 hours, from about 35% to about 95% at 18 hours, and greater than about 65% at 24 hours.
14. The oral dosage form of claim 13, providing an in-vitro release rate by weight of opioid agonist between 1% and about 50% at 1 hour, between about 2% and about 75% at 2 hours, between about 3% and about 95% at 4 hours, between about 5% and about 100% at 8 hours, greater than about 30% at 12 hours, greater than about 40% at 18 hours, and greater than about 50% at 24 hours.
15. The oral dosage form of claim 10, said dosage form providing an in-vitro release rate by weight of opioid agonist, when measured by the USP Basket or Paddle Method at 100 rpm in 900 mL aqueous buffer at a pH of between 1.6 and 7.2 at 37 0C of not less than 1% at 3 hours, not less than 2% at 6 hours, not less than 5% at 8 hour, not less
than 10% at 12 hours, not less than 15% at 18 hours, not less than 20% at 24 hours, and greater than 40% at 36 hours.
16. The oral dosage form of any of claims 1 1-15, wherein the in- vitro release rate is substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
17. The oral dosage form of any of claims 1-16, wherein the aversive agent is in the form of multiparticulates individually coated with a sequestering material which substantially prevents release of the aversive agent.
18. The oral dosage form of any of claims 1-16, wherein the agonist and aversive agent are isolated from each other in two or more distinct layers.
19. The oral dosage form of any of claims 1-16, wherein the agonist and aversive agent are interdispersed and are not isolated from each other in two distinct layers.
20. The oral dosage form of any of claims 1-16, wherein the aversive agent is dispersed in a matrix comprising a sequestering material which substantially prevents the release of the aversive agent.
21. The oral dosage form of claim 20, wherein the aversive agent dispersed in another matrix comprising the opioid agonist or is contained in a capsule with the opioid agonist.
22. The oral dosage form of any of claims 1-16, wherein the agonist and aversive agent are interdispersed and indistinguishable on the basis of physical characteristics, including color, bead diameter, density, texture, smell or flotation.
23. The oral dosage form of any of claims 1-16, wherein the aversive agent is in the form of multiparticulates coated with a sequestering material.
24. The oral dosage form of any of claims 1-16, which comprises multiparticulates in the form of inert beads coated with the aversive agent and overcoated with a sequestering material.
25. The oral dosage form of any of claims 1-16, which is in the form of a granulation comprising the aversive agent and a sequestering material.
26. The oral dosage form of any of claims 1-16, which is wherein the aversive agent is in the form of multiparticulates individually coated with a sequestering material which substantially prevents release of the aversive agent and is subsequently overcoated with the opioid agonist.
27. The oral dosage form of any of claims 1-16, which is in the form of a granulation comprising the aversive agent and the sequestering material, and the overcoated still by the opioid agonist.
28. The oral dosage form of any of claims 1-16, wherein the opioid agonist and aversive agent are in the form of a compressed tablet or a capsule.
29. The oral dosage form of any of claims 1-16, which comprises one or more opioid agonists in releasable or substantially releasable form; and one or more aversive agents in a non-releasable or substantially releasable form when said dosage form is used as intended.
30. The oral dosage form of any of claims 1-16, which comprises one or more opioid agonists in releasable or substantially releasable form, and one or more cannabinoid antagonists and one or more alcohol deterrents, each in a non-releasable or substantially non- releasable form, when said dosage form is used as intended.
31. The oral dosage form of any of claims 1-16, which comprises one or more opioid agonists in releasable or substantially releasable form, and one or more cannabinoid antagonists plus one or more alcohol deterrents, each in a non-releasable or substantially non- releasable form, when said dosage form is used as intended.
32. The oral dosage form of any of claims 1-31, wherein the amount of the opioid agonist in the claimed cannabinoid composition is from about 10 ng to about 1500 mg, more preferably, 10 ng to 1000 mg, even more preferably 0.1 mg to 800 mg, and most preferably, 0.1 mg to 500 mg.
33. The oral dosage form of any of claims 1-32, further comprising a non-opioid therapeutically active agent in releasable or substantially releasable form.
34. The oral dosage form of any of claims 1-32, further comprising one or more abuse intervention agent(s) in sequestered, partially sequestered, unsequestered, non-releasable, partially releasable or releasable form.
35. The oral dosage form of claim 34, wherein the abuse intervention agent is sequestered.
36. The oral dosage form of claims 34-35, wherein the abuse intervention agent(s) is selected from the group consisting of laxatives, cutaneous vasodilators, headache producing agents, emetics, emetogenic compound, nausea producing compounds, bittering agents, drugs that cause burning on irritation when in contact with tissue or mucous membranes (e.g., naso-mucosal irritants, oro- mucosal irritants, respiratory irritants), tissue irritants, gastrointestinal irritants, drugs that precipitate withdrawal effects, tissue dyes, lakes and colorants, beverage dyes, lakes and colorants, non-tissue staining beverage dyes, lakes and colorants (i.e., that do not stain or discolor the skin upon ingestion), fecal discolorants, urine discolorants, malodorous agents, opioid antagonists, benzodiazepine antagonists (e.g., flumazenil), and mixtures thereof.
37. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent comprises a non-toxic dye to deter surreptitious attempts at intoxication of another subject.
38. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent which comprises a non-toxic bittering agent.
39. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent which comprises a non-toxic nasal irritant to deter oral or nasal ingestion of the dosage form.
40. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent which compriss an opioid antagonist to deter oral, parenteral, nasal or inhalational use of the dosage form.
41. The oral dosage form of claim 38, wherein the abuse intervention agent is one or more bittering agents selected from the group comprising T2R or TAS2R receptor agonists, phenylthiourea (phenylthiocarbamide), natural, artificial and synthetic flavor oils, flavoring aromatics, flavoring oils, oleoresins, spearmint oil, peppermint oil, eucalyptus oil, oil of nutmeg, allspice, mace, oil of bitter almonds, menthol, citrus oils including lemon, orange, lime, grapefruit, and fruit essences, sucrose derivatives, sucrose octaacetate, chlorosucrose derivatives, quinine, denatonium, denatonium saccharide and denatonium benzoate.
42. The oral dosage form of claim 39, wherein the abuse intervention agent is one or more naso-mucosal, oro-mucosal, respiratory or tissue irritants selected from the group comprising transient receptor potential vanilloid 1 agonists, resiniferanoids, capsaicinoids, phorboid vanilloids, terpenoid 1 ,4-unsaturated dialdehydes, capsaicin, capsaicin analogs, resiniferatoxin, olvanil, piperine, zingerone, anandamide, 12- and 15-(S)-hydroperoxy-eicosatetraenoic acids, 5 ^and 15-(S)- hydroxyeicosatetraenoic acids, phorbol 12-phenylacetate 13-acetate 20-homovanillate, 2 phorbol 12,13-didecanoate 20-homovanillate, leukotriene B(4), tinyatoxin, heptanoylisobutylamide, N-(3-acyloxy-2- benzylpropy 1 )-N'-dihydroxytetrahydrobenzazepine, tetrahydro- isoquinoline thiourea analogs, heptanoyl guaiacylamide, isobutylamides, guaiacylamides, dihydrocapsaicin, homovanillyl
octylester, nonanoyl vanillylamide, formic acid, acetic acid, propionic acidy, butyric acid, valeric acid, caproic acid, caprillic acid, capric acid, oxalic acid, malonic acid, succicnic acid, glirtaric acid, adipic acid, maleic acid, fumaric acid, citric acid, sodium lauryl sulfate, poloxamer, sorbitan monoesters, glyceryl monooleates, niacin, mustard, allyl isothiocyaanate and p-hydroxybenzyl isothiocyanate and acetylsalicylic acid.
43. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent is one or more emetogenic or nausea producing agents selected from the group comprising zinc and pharmaceutically acceptable salts thereof, dopamine agonists, apomorphine, ipecac, ipecacuanha, emetine, methylcephaeline, cephaeline, psychotrine, O- methylpsychotrine, ammonium chloride, potassium chloride, magnesium sulfate, ferrous gluconate, ferrous sulfate, aloin, algarot or antimonious oxychloride, antimony trichloride, folate, folic acid, niacin and nicotinamide.
44. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent is one or more cutaneous vasodilators selected from the group comprising niacin, nicotinuric acid, beta- hydroxybutyrate and nicotinic receptor agonists, including agonists at nicotinic receptor HM74A and nicotinic receptor GPRl 09A.
45. The oral dosage form of any of claims 34-36, wherein the abuse intervention agent is one or more tissue dyes, lakes or colorants, or beverage dyes, lakes or colorants, or a beverage dye, lake and colorant that does not stain or discolor the skin upon ingestion, or a fecal discolorant or a urine discolorant.
46. The oral dosage form of any of claims 28-31 , wherein the abuse intervention agent is one or more laxatives selected from the group comprising Bis(p-hydroxyphenyl)pyridyl-2-methane, bisacodyl, bisoxatin, anthraquinone, anthraquinone analogs and derivatives (e.g., buckthorn, casanthranol, cascara, hydroxyanthracene, glucofrangulin
), dantron, danthron, docusate (e.g., docusate sodium, docusate calcium, docusate potassium), gastrointestinal chloride channel activators (e.g., chloride channel subtype 2 activators), lubiprostone, magenesium salts (e.g., magnesium citrate, magnesium hydroxide, magnesium oxide), mannitol, oxyphenisatine, polyethylene glycol, polyethylene oxide) [PEO-1500], sodium phosphate, phenolphthalein, senna, senna constituents and derivatives (e.g., sennoside A, sennoside B) and sodium picosulfate.
47. The oral dosage form of any of claims 28-31 , wherein the abuse intervention agent is one or more opioid antagonists selected from the group comprising naltrexone, methylnaltrexone, naloxone, nalmefene, cyclazocine, cyclorphan, oxilorphan, nalmefene, nadide, levallorphan, N-methylnaltrexone, N-allyllevallorphan, N-methylnaltrexone, alvimopan, N-methylnalmefene and N-allyllevallorphan.
48. The oral dosage form of claims 1-31, wherein the aversive agent is sequestered with a sequestering material selected from the group consisting of ethylcellulose, cellulose acetate, cellulose propionate (lower, medium or higher molecular weight), cellulose acetate propionate, cellulose acetate butyrate, cellulose acetate phthalate and cellulose triacetate, acetaldehyde dimethyl cellulose acetate, cellulose acetate ethylcarbamate, cellulose acetate methylcarbamate, and cellulose acetate dimethylaminocellulose acetate, acrylic acid and methacrylic acid copolymers, methyl methacrylate copolymers, ethoxyethyl methacrylates, cyanoethyl methacrylate, poly(acrylic acid), poly(methacrylic acid), methacrylic acid alkylamide copolymer, poly(methyl methacrylate), polymethacrylate, poly(methyl methacrylate) copolymer, polyacrylamide, aminoalkyl methacrylate copolymer, poly(methacrylic acid anhydride), and glycidyl methacrylate co-polymers, a poly(lactic/glycolic acid) ("PLGA"), a polylactide, a polyglycolide, a polyanhydride, a polyorthoester, polycaprolactones, polyphosphazenes, polysaccharides, proteinaceous
polymers, polyesthers, polydioxanone, polygluconate, polylactic-acid- polyethylene oxide copolymers, poly(hydroxybutyrate), polyphosphoesther and mixtures of any of the foregoing.
49. The oral dosage form of claims 1-31, wherein the aversive agent is sequestered with a sequestering material comprising a pharmaceutically acceptable biodegradable hydrophobic material.
50. The oral dosage form of any of claims 1-49, wherein the in-vitro release rate of the opioid agonist is substantially independent of pH in that a difference, at any given time, between an amount of opioid agonist released at one pH and an amount released at any other pH, when measured in-vitro using the USP Basket or Paddle Method of USP Drug Release test of U.S. Pharmacopeia (2003) at 100 rpm in 900 ml aqueous buffer, is no greater than 30%.
51. The dosage form of any of claims 1 -50, wherein the opioid agonist has agonist activity at one or more of the followings: (i) μ (mu)-opioid opioid receptor selective; (ii) δ (delta)-opioid opioid receptor selective; or (iii) K (kappa)-opioid receptor selective; and (iv) optionally, also has non-opioid receptor pharmacologic activity.
52. The dosage form of any of claims 1 to 50, wherein the opioid agonist has substantial activity at the μ (mu)-opioid receptor.
53. The oral dosage form of any of claims 1-50, wherein the opioid agonist is selected from the group consisting of alfentanil, allylprodine, alphaprodine, anileridine, benzylmorphine, bezitramide, brifentanil, buprenorphine, butorphanol, carfentanil, clonitazene, codeine, cyclorphen, cyprenorphine, desomorphine, dextromoramide, dezocine, diampromide, dihydrocodeine, dihydromorphine, dimenoxadol, dimepheptanol, dimethylthiambutene, dioxyaphetyl butyrate, dipipanone, eptazocine, ethoheptazine, ethylmeth- ylthiambutene, ethylmorphine, etonitazene, fentanyl, heroin, hydrocodone, hydroxymethylmorphinan, hydromorphone, hydroxypethidine, isomethadone, ketobemidone, levallorphan, levorphanol, levophenacylmorphan, lofentanil, meperidine,
meptazinol, metazocine, methadone, methylmorphine, metopon, mirfentanil, morphine, morphine-6-glucuronide, myrophine, nalbuphine, narceine, nicomorphine, norlevorphanol, normethadone, nociceptin/orphanin FQ (N/OFQ), normoφhine, noφipanone, ohmefentanyl, opium, oxycodone, oxymorphone, papaveretum, pentazocine, phenadoxone, phenomorphan, phenazocine, phenoperidine, pholcodine, piminodine, piritramide, propheptazine, promedol, profadol, properidine, propiram, propoxyphene, remifentanil, sufentanil, tapentadol, tramadol, trefentanil, tilidine,
54. The oral dosage form of any of claims 1-50, wherein the opioid agonist is selected from the group consisting of opioids of the phenanthrene, morphinan, benzomorphan, methadone, phenylpiperidine, propionanilide 4-anilidopiperidine, 4-aryl piperidines, and 4-Heteroarylpiperidines class.
55. The oral dosage form of any of claims 1-50, wherein the opioid agonist is in the form of a pharmaceutically acceptable salt, prodrug, ester, analog, derivative, solvate, complex, polymorph, hydrate, racemate or an individual diastereoisomers or enantiomeric isomers thereof or mixture thereof.
56. The oral dosage form of any of claims 1-50, wherein the aversive agent is selected from the group consisting of a cannabinoid antagonist, an alcohol deterrent, and mixtures thereof.
57. The oral dosage form of claim 56, wherein the aversive agent is a cannabinoid antagonist.
58. The oral dosage form of claim 56, wherein the aversive agent is an alcohol deterrent.
59. The oral dosage form of any of claims 56, wherein the aversive agent is in the form of a pharmaceutically acceptable salt, prodrug, ester, analog, derivative, solvate, complex, polymorph, hydrate, racemate or an individual diastereoisomers or enantiomeric isomers thereof or mixture thereof.
60. The oral dosage form of any of claims 56, wherein the cannabinoid antagonist is selected from the group consisting SR 141716A [Rimonabant or N-piperidino-5-(4-chlorophenyl)-l-(2,4- dichlorophenyl)-4-methyl-3-pyrazole-carboxamide]), AM251 , AM 281 ([N-moφholin-4-yl]-5-[2,4-yl]-5-[2,4-dichlorophenyl]-4-methyl- lH-pyrazole-3-carboxamide), AM630, SR 144528 ([N-[(l S)-endo- 1 ,3,3-trimethylbicyclo(2.2.1 )heptan-2-yl]5-(4-chloro-3-methyl- phenyl)-l-(4-methylbenzyl)pyrazole-3-carboxamide]), 5-(4- chloropheny I)- 1 -(2,4-dichlorophenyl)-3-hexy 1- 1 h- 1 ,2,4-triazole, 8- chloro- 1 -(2',4'-dichloropheny l)-N-piperidin- 1 -y 1- 1 ,4,5,6- tetrahydrobenzo[6,7]cyclohepta[l,2-c]pyrazole-3-carboxamide 4a, pyrazole class cannabinoid antagonists (e.g., SR141716 and SR144528), aminoalkylindole class cannabinoid antagonists (e.g., AM630), imidazolinedione class cannabinoid antagonists and triazole class cannabinoid antagonists, pyridone derivative class cannabinoid antagonists, quinolone derivative class cannabinoid antagonists, tricyclic derivatives of l-benzylpyrazole-3-carboxylic acid, HU-308, HU-210, cannabidiol, tricyclic pyrazoles, analogs of 8-chloro-l-(2',4'- dichlorophenyl)-N-piperidin-l-yl-l, 4,5,6- tetrahydrobenzo[6,7]cyclohepta[l,2-c]pyrazole-3-carboxamide, 5-(4- chlorophenyl)-l-(2,4-dichlorophenyl)-3-hexyl-lh-l,2,4-triazole.
61. The oral dosage form of claim 60, wherein the cannabinoid antagonist is CBi receptor selective.
62. The oral dosage form of claim 60, wherein the cannabinoid antagonist is CB2 receptor selective.
63. The oral dosage form of claim 60, wherein the cannabinoid antagonist has mixed CB) receptor and CB2 receptor activity.
64. The oral dosage form of claim 60, wherein the cannabinoid antagonist has activity at non-CBi and non-CB2 cannabinoid antagonist.
65. The oral dosage form of claim 60, wherein the cannabinoid antagonist is an inducer of anandamide amidase inhibitor metabolism.
66. The oral dosage form of claim 60, wherein the cannabinoid antagonist is an inducer of CBi, CB2 and non-CBi/non-CB2 cannabinoid agonist metabolism or reuptake.The oral dosage form of any of claims 1-52, wherein the ratio of the cannabinoid agonist to the aversive agent is about 1 : 100 to about 100: 1.
67. The oral dosage form of any of claims 1 -56, wherein the ratio of the cannabinoid agonist to the aversive agent is about 10: 1 to about 1 : 10.
68. The oral dosage form of any of claims 1-56, wherein the ratio of the cannabinoid agonist to the aversive agent is about 1 :5 to about 5: 1.
69. The oral dosage form of claim 58, wherein the alcohol deterrent is selected from a group consisting of disulfiram, calcium carbimide, acmaprosate, diethylthiomethylcarbamate, inhibitors of aldehyde dehydrogenase, metabolites of disulfiram, metronidazole, chlorpropamide and topiramate.
70. The oral dosage form of claim 4, wherein the ratio of the mean AUC0-t or AUCO-oo of the aversive agent after single dose oral administration of an immediate release reference product containing an equivalent amount of aversive agent to the mean AUC0-t or AUCO-cx) of aversive agent after single dose oral administration of an intact dosage form is at least 1.5: 1.
71. The oral dosage form of claim 70, wherein the ratio is at least 6: 1, or at least 10:1, or at least 20:1, or at least 30:1, or at least 40: 1, or at least 50: 1, or at least 70: 1, or at least 100:1, or at least 500: 1.
72. The oral dosage form of claim 56, wherein the amount of aversive agent included in the dosage form is from about 10 ng to 1500 mg, more preferably, 10 ng to 1000 mg, even more preferably 0.1 mg to 800 mg, and most preferably, 0.1 mg to 500 mg.
73. An oral dosage form of claim 1 wherein the abuse, misuse and tampering of the dosage form at partially blocks the "high", "liking", pleasurable, euphoric, calming, anxiolytic, mood altering, relaxing,
psychotomimetic, rewarding or reinforcing effects of the opioid agonist.
74. An oral dosage form of claim 1 wherein the abuse, misuse and tampering of the dosage form at partially blocks the "high", "liking", pleasurable, euphoric, calming, anxiolytic, mood altering, visual and perceptual altering, relaxing, psychotomimetic, rewarding or reinforcing effects of another abusable drug which is a cannabinoid agonist.
75. An oral dosage form comprising of any of claim 1 to 7, wherein the opioid agonist is in immediate release form.
76. An oral dosage form comprising of any of claim 1 to 7, said dosage form providing an in-vitro release rate by weight of opioid agonist from the dosage form of 2% to about 50% at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C.
77. An oral dosage form comprising of any of claim 1 to 7, said dosage form providing an in-vitro release rate by weight of opioid agonist from the dosage form of 2% to about 50% at one hour when measured by the USP Basket or Paddle Method at 100 rpm in 700 ml of Simulated Gastric Fluid (SGF) at 37 0C.
78. The dosage form of claims 1 to 3, where the abuse, misuse, or tampering involves attempts to liberate the opioid from the dosage form other than by oral ingestion of the intact dosage form as intended by the manufacturer or the prescribing physician.
79. The dosage form of claims 1 to 3, where the abuse, misuse, or tampering involves attempts to liberate the opioid agonist from the dosage form by use of mechanical, thermal and/or chemical means or energy to changes the physical properties of the dosage form.
80. The dosage form of claims 1 to 3, where the abuse, misuse, or tampering involves substantial solvent immersion, solvent extraction, crushing, grinding, dissolution, heating, or combustion of the dosage, followed by systemic administration of the opioid agonist or the
dosage form contents by the sublingual, buccal, transmucosal, oral, rectal, parenteral, intranasal, dermal and/or inhalational routes.
81. The dosage form of claim 33 wherein the non-opioid therapeutically active agent is an analgesic.
82. The dosage form of claim 81, wherein the analgesic is selected from the group comprising NSAIDs, NO-NSAIDs, COX-2 selective inhibitors, acetaminophen, cannbinoid agonists, nitroparacetamol, nitric oxide donors, tramadol, beta adrenergic agonists, alpha-2 agonists, selective prostanoid receptor antagonists, NO-opioid receptor agonists, local anesthetics, purinergic P2 receptor antagonists, NMDA receptor antagonists, gabapentin, pregabalin, gabapentinoids, ligands of alpha(2)delta subunits of voltage-gated calcium channels, neuronal nicotinic receptor agonists, calcium channel antagonists, sodium channel blockers, superoxide dismutase mimetics, p38 MAP kinase inhibitors, TRPVl agonists, dextromethorphan, dextrorphan, ketamine, glycine receptor antagonists, antidepressants, corticosteroids, and antiepileptics.
83. A method for treating or preventing medical conditions amenable to treatment with an opioid agonist comprising administering to a human patient in need thereof an effective amount of the oral dosage form of any of claims 1-82.
84. A method of claim 73, wherein the medical condition is Alzheimer's disease, schizophrenia, depression, alcoholism, Parkinson's disease, stroke, premature labor, endotoxic shock, hepatic cirrhosis, atherosclerosis, cancer, glaucoma, emesis, multiple sclerosis, amyotrophic lateral sclerosis, encephalitis, Huntington's disease, obesity, memory impairment, cognitive impairment, hypertension, cardiogenic shock, cerebral ischemia, myocardial infarction, neurotoxicity, febrile seizures and various intestinal disorders.
85. A method for treating or preventing pain, comprising administering to a human patient in need thereof an effective amount of the oral dosage form of any of claims 1-82.
86. A method for deterring or reducing the abuse, misuse, diversion, tampering, and/or toxicity upon tampering of abusable opioids and, optionally, co-abused cannabinoid agonists and alcohol deterrents, comprising administering to a human patient in need thereof an effective amount of the oral dosage form of any of claims 1-82.
87. Kits for use in treating or preventing diseases or disorders amenable to treatment with the dosage form of any of claims 1-82 comprising: (i) a dosage form of the invention; (ii) a container for the dosage form; and optionally, any of (iii) to (vi): (iii) a container for individual units of the dosage form (e.g., individual capsules or tablets, or blister packs); (iv) educational instructions in any media about any medical condition, its etiology, pathophysiology, consequences and treatment, potential for abuse and diversion and methods for prevention of same and information on the proper use and disposal of the medication; (v) containers or bags for the safe disposal of any remaining unused dosage form, preferably child proof and flushable; and (vi) tamper evident and child proof packaging for the kit and its contents.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US84097006P | 2006-08-30 | 2006-08-30 | |
| US60/840,970 | 2006-08-30 | ||
| US84899406P | 2006-10-04 | 2006-10-04 | |
| US60/848,994 | 2006-10-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008027442A2 true WO2008027442A2 (en) | 2008-03-06 |
| WO2008027442A3 WO2008027442A3 (en) | 2008-10-16 |
Family
ID=39136569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/019015 WO2008027442A2 (en) | 2006-08-30 | 2007-08-30 | Abuse deterrent oral pharmaceutical formulations of opioid agonists and method of use |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008027442A2 (en) |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009120735A1 (en) * | 2008-03-27 | 2009-10-01 | Us Worldmeds Llc | Composition and method for transmucosal delivery of lofexidine |
| WO2010025931A3 (en) * | 2008-09-05 | 2010-06-17 | Grünenthal GmbH | Pharmaceutical combination of 3- (3-dimethylamino-1-ethyl-2 -methyl-propyl) - phenol and an antiepileptic |
| EP2224915A1 (en) * | 2007-12-17 | 2010-09-08 | Alpharma Pharmaceuticals LLC | Pharmaceutical composition |
| EP2224806A1 (en) * | 2007-12-17 | 2010-09-08 | Alpharma Pharmaceuticals LLC | Pharmaceutical composition |
| EP2262367A2 (en) * | 2008-03-08 | 2010-12-22 | Theraquest Biosciences, Inc. | Oral pharmaceutical compositions of buprenorphine and method of use |
| US20110165248A1 (en) * | 2008-09-18 | 2011-07-07 | Meridith Lee Machonis | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| KR20110081880A (en) * | 2008-10-30 | 2011-07-14 | 그뤼넨탈 게엠베하 | Novel and potent tapentadol dosage forms |
| CN102210764A (en) * | 2011-05-30 | 2011-10-12 | 陕西师范大学 | Application of sweet almond oil in preparing medicament for treating myocardial ischemia reperfusion injury |
| CN102526353A (en) * | 2012-03-15 | 2012-07-04 | 山东阿如拉药物研究开发有限公司 | Method for preparing medicinal composition preparation for treating myocardial ischemia |
| US8460640B2 (en) | 2008-12-12 | 2013-06-11 | Paladin Labs, Inc. | Narcotic drug formulations with decreased abuse potential |
| CN103282026A (en) * | 2010-12-09 | 2013-09-04 | 欧洲凯尔特公司 | Dosage form |
| WO2014032108A1 (en) * | 2012-08-29 | 2014-03-06 | Borody Thomas J | Laxative compositions and methods for treating constipation and related gastrointestinal diseases and conditions |
| US8808745B2 (en) | 2001-09-21 | 2014-08-19 | Egalet Ltd. | Morphine polymer release system |
| US8877241B2 (en) | 2003-03-26 | 2014-11-04 | Egalet Ltd. | Morphine controlled release system |
| CN104324311A (en) * | 2014-09-22 | 2015-02-04 | 陈英娣 | Traditional Chinese medicine for treatment of multiple sclerosis |
| US9005660B2 (en) | 2009-02-06 | 2015-04-14 | Egalet Ltd. | Immediate release composition resistant to abuse by intake of alcohol |
| US9023394B2 (en) | 2009-06-24 | 2015-05-05 | Egalet Ltd. | Formulations and methods for the controlled release of active drug substances |
| WO2015071248A1 (en) | 2013-11-15 | 2015-05-21 | Synthon B.V. | Abuse-proofed extended release pharmaceutical composition comprising tapentadol |
| US9044402B2 (en) | 2012-07-06 | 2015-06-02 | Egalet Ltd. | Abuse-deterrent pharmaceutical compositions for controlled release |
| WO2015138791A1 (en) * | 2014-03-12 | 2015-09-17 | The Trustees Of Columbia University In The City Of New York | A new class of mu-opioid receptor agonists |
| US20160106737A1 (en) * | 2014-10-20 | 2016-04-21 | Pharmaceutical Manufacturing Research Services, Inc. | Extended Release Abuse Deterrent Liquid Fill Dosage Form |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| WO2016181218A1 (en) | 2015-05-13 | 2016-11-17 | Yoram Sela | Stimulant abuse-deterrent compositions |
| WO2016091805A3 (en) * | 2014-12-08 | 2016-12-15 | Develco Pharma Schweiz Ag | Naloxone monopreparation and multi-layer tablet |
| US9642809B2 (en) | 2007-06-04 | 2017-05-09 | Egalet Ltd. | Controlled release pharmaceutical compositions for prolonged effect |
| US9694080B2 (en) | 2001-09-21 | 2017-07-04 | Egalet Ltd. | Polymer release system |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| WO2018183881A1 (en) * | 2017-03-31 | 2018-10-04 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceuticals ingredients |
| US10092573B2 (en) | 2010-12-13 | 2018-10-09 | Salix Pharmaceuticals, Inc. | Gastric and colonic formulations and methods for making and using them |
| US10143706B2 (en) | 2016-06-29 | 2018-12-04 | Cannscience Innovations, Inc. | Decarboxylated cannabis resins, uses thereof and methods of making same |
| US10159268B2 (en) | 2013-02-08 | 2018-12-25 | General Mills, Inc. | Reduced sodium food products |
| US10166219B2 (en) | 2012-07-27 | 2019-01-01 | Redhill Bipharma Ltd. | Formulations and methods of manufacturing formulations for use in colonic evacuation |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10206890B2 (en) | 2008-09-05 | 2019-02-19 | Gruenenthal Gmbh | Pharmaceutical combination |
| EP3446685A1 (en) * | 2012-11-30 | 2019-02-27 | Acura Pharmaceuticals, Inc. | Self-regulated release of active pharmaceutical ingredient |
| WO2020016653A1 (en) * | 2018-07-18 | 2020-01-23 | Glatt Gmbh | Extended release formulations of cannabinoids |
| US10596211B2 (en) * | 2015-07-24 | 2020-03-24 | Shabana Naheed | Medication dispensing system |
| WO2020252284A1 (en) * | 2019-06-13 | 2020-12-17 | Imbucanna, Inc | Compressible cannabinoid pharmaceutical composition |
| WO2021062232A1 (en) * | 2019-09-26 | 2021-04-01 | The Board Of Trustees Of The Leland Stanford Junior University | Methods for reducing rewarding effects of morphine without affecting its analgesic effects |
| CN112759670A (en) * | 2020-12-17 | 2021-05-07 | 中国石油大学(华东) | Mussel bionic functionalized hydrophilic polymer and hydrophilic polymer network modified super-hydrophilic net membrane as well as preparation method and application thereof |
| CN113265436A (en) * | 2021-06-07 | 2021-08-17 | 南京中医药大学 | Method for efficient biotransformation of anthraquinone products from traditional Chinese medicine sources by using streptomyces coeruleorubidus DM |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE36547E (en) * | 1992-09-21 | 2000-02-01 | Albert Einstein College Of Medicine Of Yeshiva University | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by exogenous and endogenous opioid agonists |
| US20030004177A1 (en) * | 2001-05-11 | 2003-01-02 | Endo Pharmaceuticals, Inc. | Abuse-resistant opioid dosage form |
| US20030003113A1 (en) * | 2001-06-29 | 2003-01-02 | Lewandowski Leon J. | Individualized addiction cessation therapy |
| US20030073714A1 (en) * | 2001-08-06 | 2003-04-17 | Christopher Breder | Opioid agonist formulations with releasable and sequestered antagonist |
| US20030143269A1 (en) * | 2000-02-08 | 2003-07-31 | Benjamin Oshlack | Tamper-resistant oral opioid agonist formulations |
| US20070185145A1 (en) * | 2006-02-03 | 2007-08-09 | Royds Robert B | Pharmaceutical composition containing a central opioid agonist, a central opioid antagonist, and a peripheral opioid antagonist, and method for making the same |
-
2007
- 2007-08-30 WO PCT/US2007/019015 patent/WO2008027442A2/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE36547E (en) * | 1992-09-21 | 2000-02-01 | Albert Einstein College Of Medicine Of Yeshiva University | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by exogenous and endogenous opioid agonists |
| US20030143269A1 (en) * | 2000-02-08 | 2003-07-31 | Benjamin Oshlack | Tamper-resistant oral opioid agonist formulations |
| US20030004177A1 (en) * | 2001-05-11 | 2003-01-02 | Endo Pharmaceuticals, Inc. | Abuse-resistant opioid dosage form |
| US20030003113A1 (en) * | 2001-06-29 | 2003-01-02 | Lewandowski Leon J. | Individualized addiction cessation therapy |
| US20030073714A1 (en) * | 2001-08-06 | 2003-04-17 | Christopher Breder | Opioid agonist formulations with releasable and sequestered antagonist |
| US20070185145A1 (en) * | 2006-02-03 | 2007-08-09 | Royds Robert B | Pharmaceutical composition containing a central opioid agonist, a central opioid antagonist, and a peripheral opioid antagonist, and method for making the same |
Cited By (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8808745B2 (en) | 2001-09-21 | 2014-08-19 | Egalet Ltd. | Morphine polymer release system |
| US9694080B2 (en) | 2001-09-21 | 2017-07-04 | Egalet Ltd. | Polymer release system |
| US9707179B2 (en) | 2001-09-21 | 2017-07-18 | Egalet Ltd. | Opioid polymer release system |
| US9884029B2 (en) | 2003-03-26 | 2018-02-06 | Egalet Ltd. | Morphine controlled release system |
| US8877241B2 (en) | 2003-03-26 | 2014-11-04 | Egalet Ltd. | Morphine controlled release system |
| US9375428B2 (en) | 2003-03-26 | 2016-06-28 | Egalet Ltd. | Morphine controlled release system |
| US9642809B2 (en) | 2007-06-04 | 2017-05-09 | Egalet Ltd. | Controlled release pharmaceutical compositions for prolonged effect |
| EP2224915A1 (en) * | 2007-12-17 | 2010-09-08 | Alpharma Pharmaceuticals LLC | Pharmaceutical composition |
| EP2224806A1 (en) * | 2007-12-17 | 2010-09-08 | Alpharma Pharmaceuticals LLC | Pharmaceutical composition |
| EP2224806A4 (en) * | 2007-12-17 | 2014-02-19 | Alpharma Pharmaceuticals Llc | Pharmaceutical composition |
| EP2224915A4 (en) * | 2007-12-17 | 2014-01-22 | Alpharma Pharmaceuticals Llc | Pharmaceutical composition |
| EP2262367A4 (en) * | 2008-03-08 | 2011-04-20 | Theraquest Biosciences Inc | Oral pharmaceutical compositions of buprenorphine and method of use |
| EP2262367A2 (en) * | 2008-03-08 | 2010-12-22 | Theraquest Biosciences, Inc. | Oral pharmaceutical compositions of buprenorphine and method of use |
| WO2009120735A1 (en) * | 2008-03-27 | 2009-10-01 | Us Worldmeds Llc | Composition and method for transmucosal delivery of lofexidine |
| AU2016203053B2 (en) * | 2008-09-05 | 2018-01-25 | Grünenthal GmbH | Pharmaceutical combination |
| RU2674150C1 (en) * | 2008-09-05 | 2018-12-05 | Грюненталь Гмбх | Pharmaceutical combination |
| US10206890B2 (en) | 2008-09-05 | 2019-02-19 | Gruenenthal Gmbh | Pharmaceutical combination |
| AU2018202879B2 (en) * | 2008-09-05 | 2020-03-12 | Grünenthal GmbH | Pharmaceutical combination |
| EP2735338A1 (en) * | 2008-09-05 | 2014-05-28 | Grünenthal GmbH | Pharmaceutical combination of 3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol and pregabalin or gabapentin |
| WO2010025931A3 (en) * | 2008-09-05 | 2010-06-17 | Grünenthal GmbH | Pharmaceutical combination of 3- (3-dimethylamino-1-ethyl-2 -methyl-propyl) - phenol and an antiepileptic |
| JP2012502968A (en) * | 2008-09-18 | 2012-02-02 | パーデュー、ファーマ、リミテッド、パートナーシップ | Pharmaceutical dosage form comprising poly (ε-caprolactone) |
| US20110165248A1 (en) * | 2008-09-18 | 2011-07-07 | Meridith Lee Machonis | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| KR101456741B1 (en) * | 2008-09-18 | 2014-10-31 | 퍼듀 퍼머 엘피 | Pharmaceutical dosage forms comprising poly(e-caprolactone) |
| KR101806796B1 (en) * | 2008-10-30 | 2017-12-08 | 그뤼넨탈 게엠베하 | Novel and potent tapentadol dosage forms |
| KR20110081880A (en) * | 2008-10-30 | 2011-07-14 | 그뤼넨탈 게엠베하 | Novel and potent tapentadol dosage forms |
| JP2012507519A (en) * | 2008-10-30 | 2012-03-29 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | New, potent tapentadol dosage form |
| US9642801B2 (en) | 2008-10-30 | 2017-05-09 | Gruenenthal Gmbh | And potent tapentadol dosage forms |
| KR101712339B1 (en) | 2008-10-30 | 2017-03-06 | 그뤼넨탈 게엠베하 | Novel and potent tapentadol dosage forms |
| US8460640B2 (en) | 2008-12-12 | 2013-06-11 | Paladin Labs, Inc. | Narcotic drug formulations with decreased abuse potential |
| US9358295B2 (en) | 2009-02-06 | 2016-06-07 | Egalet Ltd. | Immediate release composition resistant to abuse by intake of alcohol |
| US9005660B2 (en) | 2009-02-06 | 2015-04-14 | Egalet Ltd. | Immediate release composition resistant to abuse by intake of alcohol |
| US9023394B2 (en) | 2009-06-24 | 2015-05-05 | Egalet Ltd. | Formulations and methods for the controlled release of active drug substances |
| CN103282026A (en) * | 2010-12-09 | 2013-09-04 | 欧洲凯尔特公司 | Dosage form |
| US10092573B2 (en) | 2010-12-13 | 2018-10-09 | Salix Pharmaceuticals, Inc. | Gastric and colonic formulations and methods for making and using them |
| CN102210764B (en) * | 2011-05-30 | 2013-04-17 | 陕西师范大学 | Application of sweet almond oil in preparing medicament for treating myocardial ischemia reperfusion injury |
| CN102210764A (en) * | 2011-05-30 | 2011-10-12 | 陕西师范大学 | Application of sweet almond oil in preparing medicament for treating myocardial ischemia reperfusion injury |
| CN102526353A (en) * | 2012-03-15 | 2012-07-04 | 山东阿如拉药物研究开发有限公司 | Method for preparing medicinal composition preparation for treating myocardial ischemia |
| US9044402B2 (en) | 2012-07-06 | 2015-06-02 | Egalet Ltd. | Abuse-deterrent pharmaceutical compositions for controlled release |
| US10166219B2 (en) | 2012-07-27 | 2019-01-01 | Redhill Bipharma Ltd. | Formulations and methods of manufacturing formulations for use in colonic evacuation |
| AU2013308403B2 (en) * | 2012-08-29 | 2019-02-07 | Salix Pharmaceuticals, Inc. | Laxative compositions and methods for treating constipation and related gastrointestinal diseases and conditions |
| WO2014032108A1 (en) * | 2012-08-29 | 2014-03-06 | Borody Thomas J | Laxative compositions and methods for treating constipation and related gastrointestinal diseases and conditions |
| AU2020210206B2 (en) * | 2012-11-30 | 2022-10-13 | Acura Pharmaceuticals, Inc. | Self-regulated release of active pharmaceutical ingredient |
| EP3446685A1 (en) * | 2012-11-30 | 2019-02-27 | Acura Pharmaceuticals, Inc. | Self-regulated release of active pharmaceutical ingredient |
| US10441657B2 (en) | 2012-11-30 | 2019-10-15 | Abuse Deterrent Pharmaceuticals, Llc | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US10688184B2 (en) | 2012-11-30 | 2020-06-23 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US11857629B2 (en) | 2012-11-30 | 2024-01-02 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceutical ingredient |
| US11540539B2 (en) | 2013-02-08 | 2023-01-03 | General Mills, Inc. | Reduced sodium food products |
| US10159268B2 (en) | 2013-02-08 | 2018-12-25 | General Mills, Inc. | Reduced sodium food products |
| US10639281B2 (en) | 2013-08-12 | 2020-05-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| WO2015071248A1 (en) | 2013-11-15 | 2015-05-21 | Synthon B.V. | Abuse-proofed extended release pharmaceutical composition comprising tapentadol |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10792254B2 (en) | 2013-12-17 | 2020-10-06 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| JP2017507960A (en) * | 2014-03-12 | 2017-03-23 | ザ・トラスティーズ・オブ・コランビア・ユニバーシティー・イン・ザ・シティー・オブ・ニューヨークThe Trustees Of Columbia University In The City Of New York | A new class of mu-opioid receptor agonists |
| US10183919B2 (en) | 2014-03-12 | 2019-01-22 | The Trustees Of Columbia University In The City Of New York | Class of mu-opioid receptor agonists |
| WO2015138791A1 (en) * | 2014-03-12 | 2015-09-17 | The Trustees Of Columbia University In The City Of New York | A new class of mu-opioid receptor agonists |
| US9707184B2 (en) | 2014-07-17 | 2017-07-18 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| CN104324311A (en) * | 2014-09-22 | 2015-02-04 | 陈英娣 | Traditional Chinese medicine for treatment of multiple sclerosis |
| US20160106737A1 (en) * | 2014-10-20 | 2016-04-21 | Pharmaceutical Manufacturing Research Services, Inc. | Extended Release Abuse Deterrent Liquid Fill Dosage Form |
| US20170020822A1 (en) * | 2014-10-20 | 2017-01-26 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| US10959958B2 (en) | 2014-10-20 | 2021-03-30 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| WO2016064873A1 (en) * | 2014-10-20 | 2016-04-28 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| WO2016091805A3 (en) * | 2014-12-08 | 2016-12-15 | Develco Pharma Schweiz Ag | Naloxone monopreparation and multi-layer tablet |
| WO2016181218A1 (en) | 2015-05-13 | 2016-11-17 | Yoram Sela | Stimulant abuse-deterrent compositions |
| US11103458B2 (en) | 2015-05-13 | 2021-08-31 | 4P-Pharma | Stimulant abuse-deterrent compositions |
| CN108135864A (en) * | 2015-05-13 | 2018-06-08 | 4P-制药公司 | The anti-abuse composition of excitant |
| US20180116961A1 (en) * | 2015-05-13 | 2018-05-03 | Yoram Sela | Stimulant abuse-deterrent compositions |
| RU2728721C2 (en) * | 2015-05-13 | 2020-07-30 | 4П-Фарма | Stimulant compositions preventing abuse thereof |
| AU2016259861B2 (en) * | 2015-05-13 | 2020-08-13 | 4P-Pharma | Stimulant abuse-deterrent compositions |
| US10596211B2 (en) * | 2015-07-24 | 2020-03-24 | Shabana Naheed | Medication dispensing system |
| US11452751B2 (en) * | 2015-07-24 | 2022-09-27 | Bao Tran | Medication dispensing system |
| US10143706B2 (en) | 2016-06-29 | 2018-12-04 | Cannscience Innovations, Inc. | Decarboxylated cannabis resins, uses thereof and methods of making same |
| US10537592B2 (en) | 2016-06-29 | 2020-01-21 | CannScience Innovations Inc. | Decarboxylated cannabis resins, uses thereof and methods of making same |
| US10383892B2 (en) | 2016-06-29 | 2019-08-20 | CannScience Innovations Inc. | Decarboxylated cannabis resins, uses thereof and methods of making same |
| WO2018183881A1 (en) * | 2017-03-31 | 2018-10-04 | Acura Pharmaceuticals, Inc. | Methods and compositions for self-regulated release of active pharmaceuticals ingredients |
| WO2020016653A1 (en) * | 2018-07-18 | 2020-01-23 | Glatt Gmbh | Extended release formulations of cannabinoids |
| US11439595B2 (en) | 2018-07-18 | 2022-09-13 | Glatt Gmbh | Immediate release formulations of cannabinoids |
| WO2020016659A3 (en) * | 2018-07-18 | 2020-03-05 | Glatt Gmbh | Multiparticulate formulations of cannabinoids |
| WO2020016658A3 (en) * | 2018-07-18 | 2020-03-05 | Glatt Gmbh | Immediate release formulations of cannabinoids |
| WO2020016656A3 (en) * | 2018-07-18 | 2020-03-05 | Glatt Gmbh | Extended release formulations of cannabinoids |
| US11918690B2 (en) | 2018-07-18 | 2024-03-05 | Glatt Gmbh | Immediate release formulations of cannabinoids |
| WO2020252284A1 (en) * | 2019-06-13 | 2020-12-17 | Imbucanna, Inc | Compressible cannabinoid pharmaceutical composition |
| WO2021062232A1 (en) * | 2019-09-26 | 2021-04-01 | The Board Of Trustees Of The Leland Stanford Junior University | Methods for reducing rewarding effects of morphine without affecting its analgesic effects |
| CN112759670A (en) * | 2020-12-17 | 2021-05-07 | 中国石油大学(华东) | Mussel bionic functionalized hydrophilic polymer and hydrophilic polymer network modified super-hydrophilic net membrane as well as preparation method and application thereof |
| CN113265436A (en) * | 2021-06-07 | 2021-08-17 | 南京中医药大学 | Method for efficient biotransformation of anthraquinone products from traditional Chinese medicine sources by using streptomyces coeruleorubidus DM |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008027442A3 (en) | 2008-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008027442A2 (en) | Abuse deterrent oral pharmaceutical formulations of opioid agonists and method of use | |
| WO2008021394A2 (en) | Pharmaceutical formulations of cannabinoids and method of use | |
| WO2008024490A2 (en) | Oral pharmaceutical formulations of abuse deterrent cannabinoids and method of use | |
| EP2448406B1 (en) | Extended release oral pharmaceutical compositions of 3-hydroxy-n-methylmorphinan and method of use | |
| US9949930B2 (en) | Opioid agonist formulations with releasable and sequestered antagonist | |
| ES2415407T3 (en) | Oral formulations of opioid agonists resistant to improper manipulations | |
| US20120178771A1 (en) | Oral Pharmaceutical Compositions of Buprenorphine and Method of Use | |
| US20140186437A1 (en) | Oral dosage forms with therapeutically active agents in controlled release cores and immediate release gelatin capsule coats | |
| US20140099376A2 (en) | Sequestered antagonist formulations | |
| US20100249045A1 (en) | Multimodal Abuse Resistant and Extended Release Opioid Formulations | |
| HUE028041T2 (en) | Tamper-resistant products for opioid delivery | |
| WO2012058695A2 (en) | Compositions of (-)-17-(cyclobutylmethyl) morphinan-3, 14-diol | |
| EP3169316A1 (en) | Compositions and methods for reducing overdose | |
| EP2405754A1 (en) | Modified release pharmaceutical compositions of buprenorphine | |
| AU2013200828A1 (en) | Oral dosage forms with therapeutically active agents in controlled release cores and immediate release gelatin capsule coats |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07837500 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| NENP | Non-entry into the national phase in: |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07837500 Country of ref document: EP Kind code of ref document: A2 |